CellNavi方法细节:预测指导细胞状态转移的基因
在细胞状态转分化过程中,仅有少数特定基因发挥关键驱动作用。然而,找到参与不同转分化过程的关键基因并非易事。为此,本研究提出一种基于深度学习的框架——CellNavi,旨在预测驱动细胞状态转分化的基因。CellNavi在细胞状态流形的基础上构建驱动基因预测器,该细胞状态流形通过学习大规模高维转录组数据,并整合具有方向连接的基因图,从而捕捉细胞的固有特征。
实验分析表明,在不同细胞类型、条件及研究中,对于由遗传扰动、化学扰动和细胞因子扰动诱导的细胞转分化,CellNavi均能准确预测其驱动基因。借助具有生物学意义的细胞状态流形,CellNavi在细胞分化、疾病进展、药物响应等关键转分化相关任务中表现出色。CellNavi在驱动基因预测和细胞状态调控领域取得了重大突破,为疾病生物学研究和治疗方法开发开辟了新路径。
来自:CellNavi predicts genes directing cellular transitions by learning a gene graph-enhanced cell state manifold,Nature Cell Biology
链接:https://www.nature.com/articles/s41556-025-01755-1
目录
- 背景概述
- 方法概述
- 方法-细节
- 输入嵌入
- 基因名称嵌入
- 基因表达嵌入
- 细胞流形模型CMM
- CMM 预训练
- 驱动基因预测
- 先验基因图
背景概述
理解细胞转分化的遗传驱动因子,对于阐明复杂的生物学过程和疾病机制至关重要。然而,识别这些驱动因子本质上具有挑战性:一方面,参与细胞转分化的基因数量庞大且相互作用复杂;另一方面,实验研究能力有限且生物学知识尚不完全,两者形成鲜明对比。因此,开发能够在不同场景下预测驱动基因的计算机模拟(in silico)方法具有重要意义。
传统上,确定关键驱动基因的研究工作主要依赖于基于网络的方法,其中尤其以基因调控网络(GRN)为核心。尽管以 GRN 为中心的研究方法已取得显著进展,但它们仍存在局限性,阻碍了其更广泛的应用。例如,在异质性细胞群体中推导准确的 GRN 仍是一项挑战。此外,GRN 模型往往更优先关注转录因子,可能会忽略细胞转分化过程中的非转录驱动因子。这一局限使得我们对疾病进展、免疫调节和药物反应等复杂细胞过程的理解受到限制。
CellNavi 是一种旨在预测驱动基因并解析细胞转分化过程的深度学习框架。CellNavi 在一个经过学习的流形(该流形可对有效细胞状态进行参数化)基础上,构建了驱动基因预测器(DGP)。构建该流形的方法是:将原始细胞状态表征映射到低维坐标空间,其中坐标维度对应细胞状态的固有特征,空间距离则反映细胞间的生物学相似性。为构建这一流形,CellNavi 以大规模高维单细胞转录组数据为训练基础,同时结合能揭示细胞状态潜在结构的先验有向基因图。通过将细胞数据投影到这一低维且生物学关联性更强的有意义空间中,CellNavi 形成了一个具有通用性的框架,可在不同细胞背景下实现泛化应用 —— 即便在以往未研究过的细胞类型或条件下,也能稳定地预测驱动基因。
研究结果表明,CellNavi 在多种生物学转分化过程中均擅长预测驱动基因,在永生化细胞系和原代细胞两类研究对象的定量任务中,均展现出优异性能。该框架能够识别 T 细胞分化过程中的关键调控因子,还能挖掘与神经退行性疾病相关的核心基因。值得注意的是,CellNavi 无需针对特定药物进行训练,就能推断出药物化合物的作用机制,这凸显了其在药物研发领域的应用潜力。综上,CellNavi 为解析细胞状态转分化及其潜在机制提供了一个强有力的框架,对推动细胞生物学研究和疾病研究具有深远的应用前景。
方法概述
CellNavi 旨在为特定的细胞转分化过程预测驱动基因,其中源细胞和目标细胞的转录组数据分别代表这些转分化过程的初始状态和终末状态(图 1a–c)。
CellNavi 包含两个主要组件:一是细胞流形模型(CMM),负责捕捉并表征细胞状态;二是驱动基因预测器(DGP),可基于学习到的细胞表征识别驱动这些转分化过程的关键基因(图 1b)。
CMM 的设计目的是捕捉不同生物学背景下的有效细胞状态。尽管转录组常被用于表征细胞状态,但有效细胞状态并非遍布整个高维转录组空间,而是形成一个低维流形(图 1c)。为对这一特征进行建模,CMM 会将转录组向量映射到低维坐标空间 —— 该空间可表征细胞状态的固有特征,同时能保留细胞间的相对相似性。
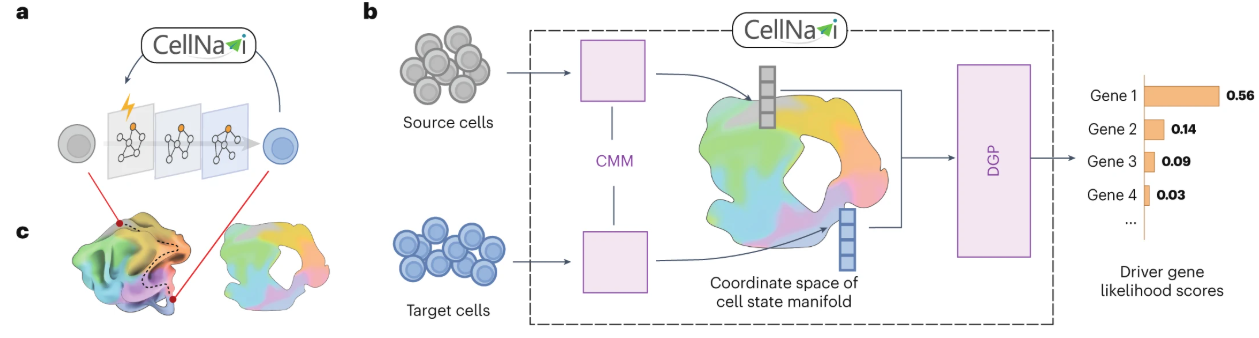
- 图1a:CellNavi 任务的概念示意图。给定一对在刺激诱导下发生转分化的源细胞与目标细胞,CellNavi 会预测导致该转分化过程的驱动基因。
图1b:CellNavi 的工作流程。细胞流形模型(CMM)将源细胞和目标细胞映射到细胞流形的坐标空间中,随后驱动基因预测器(DGP)利用 CMM 生成的细胞坐标,依据可能性分数对候选基因进行排序。
图1c:细胞流形及其坐标空间的示意图。
作者首先整理了一个包含约 2000 万单细胞转录的数据,该数据集来源于人类细胞图谱(HCA,图 1d);同时,作者采用了一种基于注意力机制的 Transformer 架构(这类架构因其能识别大规模数据中的复杂模式而闻名),用于训练细胞流形模型(CMM,图 1e)。CMM 的训练涉及一项自监督下采样重建任务。
为优先获取细胞层面而非基因层面的表征,作者开发了一个解码器模块:该模块可从 CMM 生成的细胞坐标(即细胞在细胞状态流形坐标空间中的表征)中重建基因表达谱(图 1e 及扩展数据图 1)。这种方法能够对齐不同测序深度下的细胞,并重现单细胞的发育轨迹,这表明该方法既能捕捉细胞内特征,也能捕捉细胞间特征。
然而,仅依赖转录组数据可能会忽略基因间复杂的相互作用 —— 而这种相互作用对于描述和区分细胞状态至关重要。为解决这一问题,作者将 2000 万个细胞特异性基因图整合到 CMM 的训练过程中(图 1d、e)。这些基因图对有向连接进行了编码,而这些有向连接源自一个涵盖 3 万多个人类基因及其相关信号通路的先验网络(NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020))。更具体地说,在这些基因图中,每条边代表两个基因之间的因果关系,边的方向则表明一个基因对另一个基因的调控作用。
这些基因图提供了有关基因间复杂依赖关系的更丰富信息,这种信息超越了简单的转录组数据,因此能更好地揭示有效细胞空间中潜在的内在变量。为充分利用这些基因图,作者将 CMM 中的标准 Transformer 编码器层替换为基因图注意力层(扩展数据图 1b)。这类图层的设计灵感来源于为图数据定制的注意力机制变体,能够处理基因网络,从而使模型能够整合关键的基因间关系。通过这些设计,模型得以构建一个能系统表征细胞状态、有效反映细胞间关系的流形,为驱动基因预测奠定了信息丰富的基础。

- 图1d:用于细胞流形模型(CMM)训练的数据。exps 为 “experiments”(实验)的缩写。
- 图1e:细胞流形模型(CMM)的训练过程。CMM 包含 6 个基因图注意力(attn.,即 attention)层,这些层旨在整合基于图的信息。训练期间,会从整理好的人类细胞图谱(HCA)数据集中随机抽取单细胞转录组谱作为输入,模型生成的细胞嵌入随后会被 Transformer 解码器用于重建基因表达谱。

- 扩展图1a:细胞流形模型(CMM)旨在借助由基因图注意力层(GeneGraph Attention Layers)构成的 Transformer 变体,实现基因表达谱的重建。

- 扩展图1b:基因图注意力层(GeneGraph Attention Layer)通过其多头注意力层整合先验基因图。需注意的是,仅使用输入样本中包含节点(即所有非零表达基因)的基因子图进行注意力计算。
在该流形的基础上,作者开发了驱动基因预测器(DGP),用于预测驱动特定细胞转分化过程的基因(扩展数据图 3a)。DGP 以CRISPR筛选数据为训练数据,这类数据可将遗传扰动与随之发生的细胞状态变化关联起来。作者将未受扰动的对照细胞和经 CRISPR 扰动的细胞分别指定为源细胞对和目标细胞对,并将经验证的扰动基因作为标签,用于细胞流形模型(CMM)与 DGP 的联合训练(微调)(图 1f)。具体而言,对于每一对细胞,其转录组谱会通过 CMM 转化为细胞坐标,随后这些坐标会被 DGP 处理,生成一个可能性分数向量 —— 该向量可指示各类候选基因调控该转分化过程的概率(扩展数据图 3a)。

- 扩展图3a:驱动基因预测器(DGP)会对细胞流形模型(CMM)输出的细胞坐标对进行拼接处理,通过生成可能性分数向量来预测驱动基因。模型中还包含一个可选的判别器,用于对齐训练数据与测试数据,以确保预测结果的一致性和准确性。

- 图1f:用于DGP训练的数据。
- 图1g:CellNavi 的应用场景与测试案例。MoA 为 “mechanism of action”(作用机制)的缩写。
研究表明,CellNavi 经 CRISPR 筛选数据(这类数据通常基于培养细胞或均质细胞群体获取,聚焦于即时遗传扰动)微调后,可推广应用于异质性组织和原代细胞中更复杂的转分化过程(图 1g 及扩展数据图 3b)。借助具有生物学意义的流形,CellNavi 能将从 CRISPR 筛选中获得的知识推广到其原始应用范围之外,覆盖那些常规 CRISPR 方法难以研究的细胞转分化过程。
然而,作者也承认,在特定场景下,若能利用相关 CRISPR 数据集对 CellNavi 进行额外微调,其性能可能会进一步提升。整合更多实验数据,或许能在最小化模型适配工作的前提下,进一步增强其在不同生物学场景中的适用性。
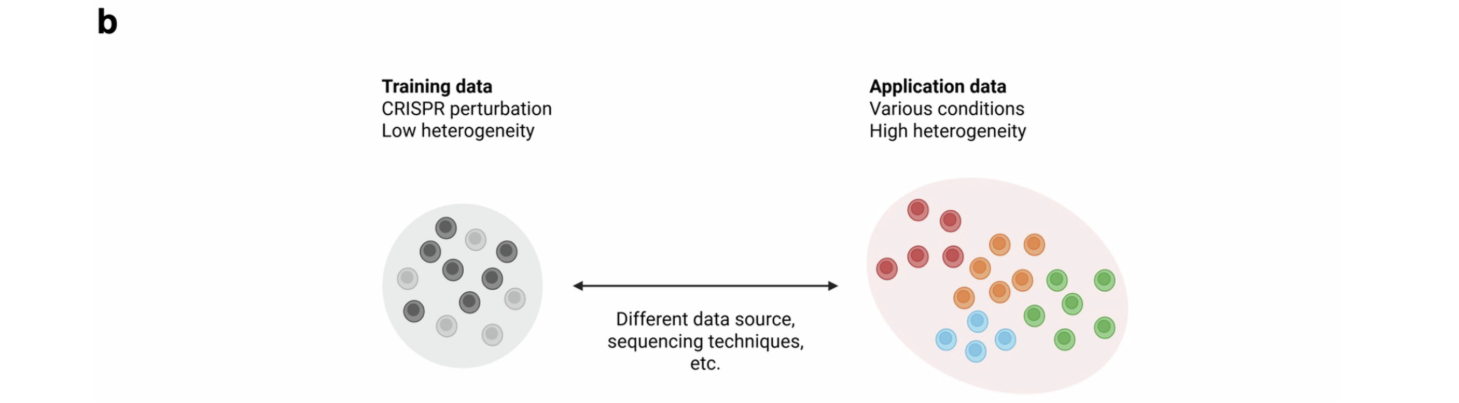
- 扩展图3b:微调数据(左侧)由异质性较低的 CRISPR 扰动数据集构成,这类数据通常来源于变异性有限的受控实验。应用数据(右侧)则涵盖了异质性较高的多种生物学条件,包括不同的细胞类型、扰动类型以及测序平台。双向箭头代表了将模型从微调场景推广到实际应用场景时,由数据源差异、测序技术差异及生物学变异性所带来的挑战。
方法-细节
输入嵌入
在CellNavi中,作者使用单细胞原始计数矩阵作为唯一的输入。具体来说,scRNA-seq被处理成X∈RN×GX\in\R^{N\times G}X∈RN×G,其中每个元素Xn,gX_{n,g}Xn,g表示细胞nnn中基因ggg的表达量。为了更好地描述基因在细胞中的状态,作者在其输入嵌入中同时涉及基因名称和基因表达信息。形式上,token的输入嵌入是基因名称嵌入和基因表达嵌入的串联。
基因名称嵌入
作者在 CellNavi 中使用可学习的基因名称嵌入。基因的词汇表是通过在所有数据集中获取基因名称的并集来获得的。然后,将词汇表中每个基因的整数标识符输入嵌入层,以获得其基因名称嵌入。此外,还合并了一个标记CLS在词汇表中,用于把所有基因聚合到一个细胞表示中。细胞nnn的基因名称嵌入表示为hn(name)∈R(G+1)×Hh_{n}^{(name)}\in\R^{(G+1)\times H}hn(name)∈R(G+1)×H:
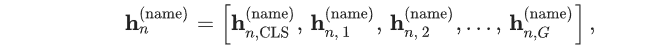
其中,HHH是嵌入的维度,设置为256。
基因表达嵌入
基因表达建模的一个主要挑战是不同测序方案之间绝对幅度的差异,作者通过使用移位对数对每个细胞的原始计数表达式进行归一化来应对这一挑战,该对数定义为:
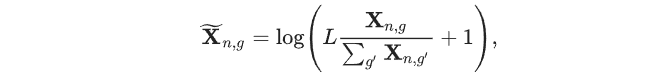
其中,LLL是一个缩放因子,作者使用了一个固定值L=1×104L=1\times 10^{4}L=1×104,然后,在归一化后的计数X~n,g\widetilde{X}_{n,g}Xn,g上应用线形层获得基因表达嵌入。对于CLS token,作者将其设置为基因表达嵌入的唯一值。细胞nnn的基因表达嵌入表示为hn(expr)∈R(G+1)×Hh_{n}^{(expr)}\in\R^{(G+1)\times H}hn(expr)∈R(G+1)×H:
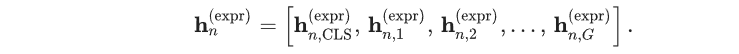
细胞nnn的最终嵌入定义为:
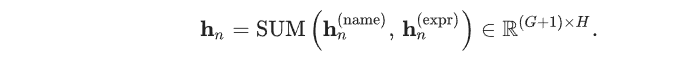
细胞流形模型CMM
CMM 由6层 Transformer 变体组成,专为处理图结构数据(GeneGraph 注意力层)而设计。编码器采用输入嵌入来生成细胞表示,并仅使用非零表达的基因。为了进一步加快训练速度,也作为一种数据增强方法,作者执行了一种基因采样策略,随机选择最多 2,048 个基因作为输入。应该注意的是,该策略仅在训练期间应用。所有非零基因都包含在推理阶段,以避免信息丢失。作者使用hn(l)h_{n}^{(l)}hn(l)表示细胞nnn在第lll层的嵌入:
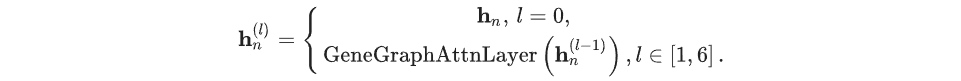
每个 GeneGraph 注意力层中的多头注意力模块由三个组件组成。除了自注意力模块外,还加入了中心性编码模块和空间编码模块,以修改用于图数据集成的标准自注意力模块。
首先介绍标准的自注意力模块。令NheadsN_{heads}Nheads为注意力模块中的头数。在第lll层,头iii中,自注意力计算为:
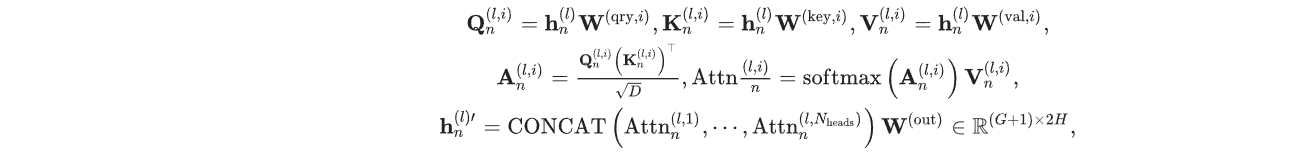
其中,W(qry,i)W^{(qry,i)}W(qry,i),W(key,i)W^{(key,i)}W(key,i),W(val,i)∈R2H×DW^{(val,i)}\in\R^{2H\times D}W(val,i)∈R2H×D是投影细胞nnn输入嵌入hn(l)h_{n}^{(l)}hn(l)的可学习矩阵,将输入嵌入投影为Qn(l,i)Q_{n}^{(l,i)}Qn(l,i),Kn(l,i)K_{n}^{(l,i)}Kn(l,i),Vn(l,i)V_{n}^{(l,i)}Vn(l,i),W(out)∈R(DNheads)×2HW^{(out)}\in\R^{(DN_{heads})\times 2H}W(out)∈R(DNheads)×2H是精细化多头注意力输出的可学习线性投影,DDD是每个注意力头的特征维度。多头注意力的输出hn(l)′h_{n}^{(l)'}hn(l)′然后通过Layer Norm层和多层感知器 (MLP) 模型,产生最终输出hn(l+1)h_{n}^{(l+1)}hn(l+1)作为下一层的输入。
标准注意力机制独立处理每个基因的特征,而基因图则包含基因之间的关系信息。为了将基因图信息纳入模型,中心性编码模块(centrality encoding module)将关系信息投射到每个单个基因的调控活动特征中,空间编码模块直接将关系信息与注意力机制相结合。具体地,作者定义zdeg−(G,g)−z^{-}_{deg^{-}(G,g)}zdeg−(G,g)−和zdeg+(G,g)+z^{+}_{deg^{+}(G,g)}zdeg+(G,g)+,这是描述基因图GGG中基因 g 的入度(deg⁻)和出度(deg⁺)的可学习嵌入向量。作者将这些嵌入向量添加到基因嵌入向量中,以更新细胞编码:
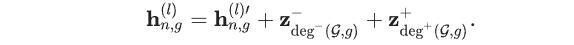
由中心性编码模块(centrality encoding module)执行的此次细胞编码更新,会在自注意力模块(self-attention module)之前完成(扩展图1b)。
空间编码模块(spatial encoding module)旨在从基因图中捕捉基因间的调控关系。为此,作者生成了距离矩阵S∈NG×GS\in\N^{G\times G}S∈NG×G,该矩阵包含基因图GGG中基因对之间的最短距离。作者将矩阵SSS中的每个元素作为可学习偏置项,添加到注意力权重中,具体(公式)如下:
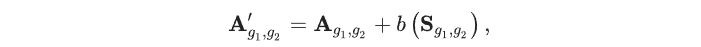
其中,bbb是可学习的标量函数,输入为距离Sg1,g2S_{g_1,g_2}Sg1,g2。作者使用A′A'A′替代原始注意力AAA,CMM输出的细胞表示为hn,CLS(6)h_{n,CLS}^{(6)}hn,CLS(6),随后通过一个全连接层,其维度从256转为2048,此结果作为流形中细胞nnn的坐标(coordinate),记为CRDnCRD_{n}CRDn。
CMM 预训练
细胞流形模型(CMM)旨在生成能够对细胞状态的固有特征与变量(其数量远少于原始基因表达谱表征的维度)进行参数化的细胞坐标,并在向量空间中保持细胞间的相似性,从而为驱动基因预测器(DGP)提供简洁且具有生物学意义的输入表征。为实现这一目标,作者设计了一项下采样重建预训练任务:该任务要求 CMM 基于某一细胞(记为细胞nnn)的下采样基因表达数据(记为Xn(ds)X_{n}^{(ds)}Xn(ds))生成细胞坐标,且该坐标需能让一个独立的解码器模型尽可能准确地重建出该细胞的原始基因表达数据(记为XnX_{n}Xn)。通过这一设计,CMM 被促使捕捉原始基因表达各维度间的共变模式,进而助力其提取细胞状态潜在的固有变量。
具体而言,在该下采样过程中,作者通过二项分布对每个基因的原始计数表达量进行下采样处理。第 nnn 个细胞中第 ggg 个基因的下采样表达量(记为Xn,g(ds)X_{n,g}^{(ds)}Xn,g(ds) )通过以下(方式 / 公式)生成:
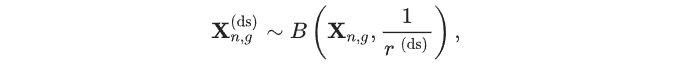
其中,r(ds)r^{(ds)}r(ds)是均匀采样的下采样率,BBB表示二项分布。解码器是两个线性层组成的MLP。对于每个下采样的基因表达,解码器会连接细胞坐标CRDnCRD_{n}CRDn,MLP输出为XnX_{n}Xn的相同形状。重建目标为:
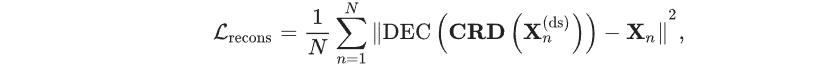
CMM和解码器都会进行优化,预训练后,CMM将用于驱动基因预测,解码器被丢弃。
驱动基因预测
驱动基因预测器(DGP)是由两个线性层组成的MLP。它经过优化,可从 CMM 输出的一对细胞坐标中预测受扰动的基因。更具体地说,源细胞的转录组XsrcX_{src}Xsrc和目标细胞XtgtX_{tgt}Xtgt映射到细胞坐标CRDsrcCRD_{src}CRDsrc和CRDtgtCRD_{tgt}CRDtgt。对于直接输入特征,DGP连接两个细胞坐标,然后进行MLP,MLP输出基因的logits,使用CE训练DGP:
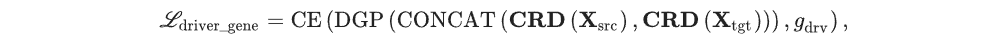
在训练DGP的时候,CMM也会更新参数。
先验基因图
先验基因图谱由NicheNet构建,其中GRN和细胞信号网络被整合。基因图是一个方向图。更具体地说,对于图上的每个基因节点,传入边的数量对应于调节它的基因,而传出边的数量代表它调节的基因。如果两个基因在任一单独的网络中链接,则它们之间建立了联系。由此产生的综合图具有 33,354 个基因,每个基因都由一个独特的人类基因符号表示,并包括 8,452,360 条表示潜在相互作用的边。CellNavi中使用了 NicheNet 网络的未加权版本。对于每个细胞,去除单细胞转录组学谱的原始计数矩阵中值为0的基因节点,以构建细胞类型特异性基因图。在预训练期间,当对单细胞转录组进行下采样时,仅保留作为模型输入包含的非零基因,以生成指导模型任务的样本特定图。
为了评估图连通性和结构的影响,作者生成了替代图配置,如下所示:
- 1)全连通图:最大连通图,其中每对基因都由权重相等的边连接。
- 2)稀疏图:通过将原始图中的边总数下采样到其总边的 1/10 和 1/20 来创建图形,从而能够评估连接性降低对性能的影响。
- 3)随机图:生成随机图的同时保留了原始图的节点数量和某些结构属性,例如自循环。边是以概率方式引入的,以保持与原始图的稀疏性和连通性的整体一致性。