玉米籽粒品质相关性状的GWAS和Meta-QTL分析

https://www.mdpi.com/2223-7747/13/19/2730
4. 材料和方法
4.1. 材料和实验设计
本研究分析了中国北京市农业科学院提供的 368 种材料中的 260 个玉米自交系,这些玉米自交系表现出优异的适应性。2020 年,位于中国甘肃省武威市黄阳实验站(北纬 37.97°,东经 102.63°)的玉米育种试验田被用于种植。实现了一个完全随机的区组设计,由三个重复组成。每个玉米自交系种植成两行,每行长 4 m,株距 0.25 m,行距 0.4 m。田间管理实践符合当地农业生产标准。
4.2. 表型性状的确定
收获后,测量与籽粒相关的品质性状。使用 NIRS DA 1650™ (Foss, Hilleroed, Denmark) 测量玉米粒的蛋白质含量 (PC)、油含量 (OC) 和淀粉含量 (SC)。所有结果均以干基 (%) 报告,每个样品重复 3 次,平均值用于统计分析。
4.3. 表型数据分析
使用 IBM SPSS Statistics 21 和 Origin 2022 软件对表型数据进行描述性统计分析和相关性分析,并生成相应的图表。
4.4. 全基因组关联研究
对分布在整个玉米基因组中的 558,529 个 SNP 位点进行全基因组关联分析,最小等位基因频率为 ≥0.05。基因型数据可在网站上访问(http://www.Maizego.org/Resources.html,2024 年 6 月 1 日访问)[10]。对基因进行质量控制,并使用 Tassel 5.0 软件中的混合线性模型 (MLM) 方法进行 GWAS 分析。根据其分位数-分位数 (QQ) 散点图选择最佳模型,对每个性状进行 GWAS 分析。Bonferroni 阈值是使用 R 语言确定的,显着性阈值设置为 p = 1 × 10−5用于识别重要的 SNP。QQ 图是使用 R 中的“CMplot”包生成的。
4.5. 候选基因挖掘和功能分析
以前的研究人员利用大约 560,000 个 SNP 来评估该组内连锁不平衡 (LD) 衰减的程度,揭示了相关组的 LD 衰减距离为 50 kb (R2= 0.1)。因此,本研究采用该衰减距离作为总体的 LD 衰减距离。这是基于玉米 B73 基因组序列的上游和下游显著相关的 SNP 标记的物理位置,RefGen_v3总跨度为 100 kb。利用 NCBI (https://www.ncbi.nlm.nih.gov/) 和 MaizeGDB (https://www.maizegdb.org/) 的资源,我们搜索了与每个性状相关的所有候选基因,并根据其功能注释选择了最合适的基因作为候选基因。来自各种玉米组织的转录组数据从 qTeller 网站下载(https://qteller.maizegdb.org/,2024 年 6 月 1 日访问)。这个全面的数据集包括28个组织和发育阶段[40],如胚胎(16、18、20、22、24 DAP)、胚乳(12、16、18、20、22、24 DAP)、全种子(2、4、6、8、10、12、14、18、20、22、24 DAP)、花药(R1)、玉米棒(R1、V18)、蚕丝(R1)和穗原基(2-4 mm和6-8 mm)。该数据集有助于对候选基因进行组织特异性表达分析。
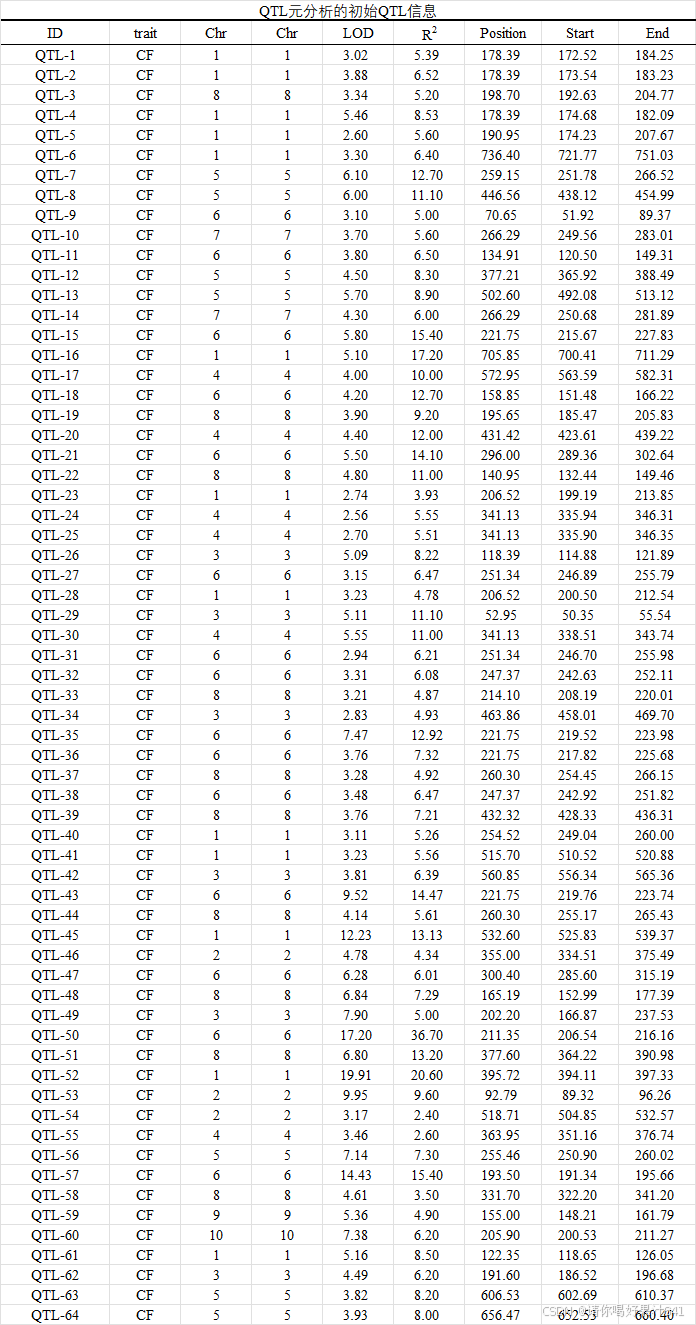
4.6. 玉米品质性状相关 QTL 信息的集合
本研究利用 Web of Science (http://www.webofknowledge.com/) 和中国国家知识基础设施 (https://www.cnki.net/) 收集有关玉米品质性状相关数量性状位点 (QTL) 的信息。于 2024 年 6 月 1 日访问,使用 “玉米”、“质量”、“淀粉”、“油”、“蛋白质”、“QTL” 和 “高密度遗传图谱” 等关键词进行搜索,确定了 2005 年至 2024 年间发表的 20 多篇相关文献。选择提供 QTL 区间和侧翼标记的文章进行编译(表 S8)。对于具有全面遗传图谱和 QTL 信息的实验,根据软件的格式化要求对数据进行组织。这包括所有基本参数,例如 QTL 名称、位置、性状、连锁组、LOD(赔率似然)值、置信区间 (CI) 和解释的表型方差 (R2).此外,从 MaizeGDB 网站(https://maizegdb.org/data_center/map?id=1140201,2024 年 6 月 1 日访问)下载了 IBM2 2008 Neighbors 地图中的信息,作为统一的遗传和参考地图。
4.7. QTL 信息的整合
使用 BioMercator 4.2.3 软件进行 QTL 预测和荟萃分析 [41]。置信区间 (CI) 和 R2的 QTL 是两个关键参数。QTL 的 Meta 分析主要依赖于 QTL LOD 评分 R2、position 和 CI 一起。在收集的QTL数据缺乏95%置信区间的情况下,根据Darvasi和Soller [42]提供的公式进行推断,其中N代表原始作图群体的大小。
CI = 530/(N × R2) (1)
CI = 163/(N × R2) (2)
公式 (1) 适用于回交和 F2映射群体,而公式 (2) 适用于 RIL 映射群体。
4.8. QTL 预测和荟萃分析
应通过遗传数据加载上传完整的遗传图谱和收集的 QTL 信息,然后将 QTL 映射到参考图谱。IBM2 Neighbors 的遗传连锁图谱整合了玉米高密度分子标记连锁图谱 (Intermated B73 × Mo17 Map;IBM) 和其他分子标记物连锁图谱。这张综合图谱包括 19,111 个基因座,其中包括 RFLP、SSR 和 RAPD 标记,以及基因和序列探针,总长度为 7898.35 cM(来源:https://maizegdb.org/data_center/map,2024 年 6 月 10 日访问;最后更新于 2022 年 8 月 10 日,由 Marty Sacks 提供)。 对每条染色体上存在的 QTL 簇进行 MQTL 分析。在该分析中,采用各种标准——AIC (Akaike 信息标准)、AICc (AIC 校正)、AIC3 (AIC3 候选模型)、BIC (贝叶斯信息标准)和 AWE (证据平均权重)——来评估所有潜在的 QTL 组合。表现出最低 AIC 值的模型表明存在多个 QTL,用于元 QTL 分析的初始 QTL 数量至少设置为 3 [43]。此外,计算 95% QTL 的位置和置信区间 (CI),并从 MaizeGDB 中鉴定 QTL 的侧翼标记。从软件获得 MQTL 结果后,使用 MaizeGDB 确定 2008 年 IBM2 邻居的 B73 (AGPv 3) 上相应标记的物理位置。最后,还从 MaizeGDB 中检索到与鉴定的 QTL 相关的候选基因。