MR-link-2:多效性顺式孟德尔随机化分析!
生信碱移
多效性鲁棒MR
孟德尔随机化能够从观测数据中识别因果关系,但当遗传工具局限于单一关联区域时,其第一类错误率会升高。一项研究提供了MR-link-2方法,能够从单一关联区域的汇总统计数据中识别出具有多效性鲁棒性的因果关系,使其特别适用于分子表型的应用研究
在人类生物医学研究中,确定因果关系的金标准是随机对照试验(RCT),但其高成本、伦理限制和实施难度,使得许多生理或分子层面的因果机制难以通过实验手段直接检验。孟德尔随机化(MR)作为一种观察性因果推断方法,通过将基因变异当作天然随机试验工具,已成功揭示了低密度脂蛋白胆固醇(LDL‑C)与冠心病、酒精摄入与心血管疾病等经典因果关系。
不了解基础概念的铁子,可以点击阅读下面的文章:《一文读懂孟德尔随机化》。
传统MR方法多借助数十至数百个全基因组上的独立工具位点,期望不同位点的偏倚相互抵消。但对分子表型(比如蛋白质丰度、代谢物浓度等),这种多位点汇总策略往往无法满足工具数量需求,因为这些分子性状通常只有单一区域或少数区域存在显著遗传信号(并且一般是顺式元件,即在相应基因坐标附近)。目前,对分子表型因果关系的鉴定主要依赖最邻近基因分析(如MAGMA)或共定位分析等方法。这些方法虽能定位相关分子,却并不严格检验两性状间的因果性。举个例子,共定位只能说明两个性状共享同一因果变异,但无法区分直接因果或受共同混杂因素驱动。
早在2020年,来自意大利的研究者便提出了MR-link工具,专门用于整合顺式信息挖掘分子表型的遗传关联。就在2025年7月3日,他们又在Nature Communications [IF:15.7]发表了该工具的进阶版MR-link2,该工具将原版方法中对个体数据输入依赖转换为对公开汇总统计和LD参考面板的输入。这样子既保留了对多效性与局部遗传架构的建模,又大幅提升了方法在更多数据上的通用性。

DOI: 10.1038/s41467-025-60868-1。
简单来讲,MR‑link‑2的核心是其在使用单一区域的情况下,不再仅依赖少数显著SNP作为工具变量,而是将该区域内所有SNP及它们的连锁不平衡(LD)矩阵一起纳入一个联合似然模型。这个模型中既包含SNP对暴露X的局部遗传贡献,也显式设立一个多效性方差参数σ²ₚ 来捕获SNP对结局Y的直接或其他路径影响,最后通过似然比检验分离并估计真正的因果效应α,从而在校正多效性干扰后严格控制假阳性率。
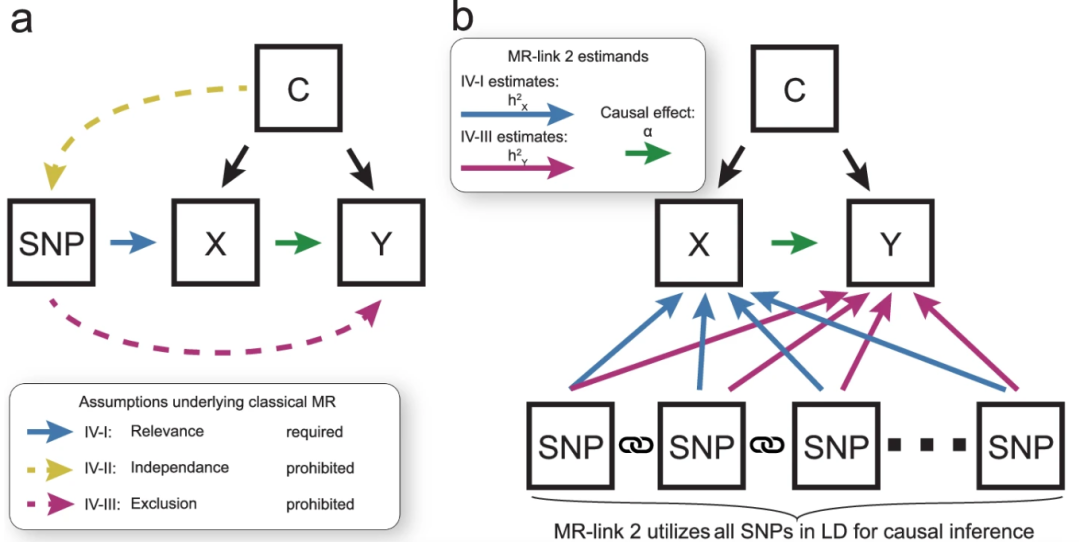
图:传统MR(左)与MR‑link‑2(右)的建模思路。a.传统MR仅选取一个或少数显著SNP作为工具,需要假定它们只通过暴露X影响结果Y(蓝色箭头),而与混杂因素C独立(黄色虚线禁止)且不直接作用于Y(紫色虚线禁止)。一旦这些假设被违背,就会产生假因果;b.MR‑link‑2则将一个基因区域内的所有SNP及其相互之间的连锁不平衡一并纳入联合似然模型,蓝色箭头估计SNP→X的局部遗传效应,紫色箭头估计SNP→Y的多效性偏倚,并在此基础上提取绿色箭头所示的真实因果效应α,从而在仅有单一区域信号的情况下依然能够有效剔除多效性干扰、严格控制假阳性率。
本文简要介绍MR‑link‑2使用,感兴趣的铁子可以进一步阅读:
-
https://github.com/adriaan-vd-graaf/mrlink2
0.环境配置
MR‑link‑2提供了python版本的命令行实现,作者建议使用python大于等于3.6。在此基础上,需要安装一些必要的py库:
pip3 install numpy scipy pandas bitarray pytest duckdb pyarrow
同时,确保该环境能够调用plink1.9软件,可以通过下述代码打印出路径,验证是否配置正确:
which plink
01.输入文件格式示例
MR‑link‑2需要提供两份GWAS汇总统计和一组PLINK基因型文件的前缀名称(.bed/.bim/.fam后缀)。其中,两份GWAS汇总统计数据分别是研究的对象:
-
暴露文件(
sumstats_exposure.txt) -
结局文件(
sumstats_outcome.txt)
#暴露文件sumstats_exposure.txt
pos_name chromosome position effect_allele reference_allele beta se z pval n_iids
rs11694978 2 102000210 A G 0.154273401479263 0.013972828758673157 11.040956999025918 2.424371554145676e-28 5000
rs62156072 2 102000790 A G -0.015436749281195056 0.014140450535761791 -1.0916730865225879 0.27497681883818303 5000
rs111408528 2 102001310 T C -0.015436749281195056 0.014140450535761791 -1.0916730865225879 0.27497681883818303 5000
rs182612067 2 102001874 T C 0.03768964658579835 0.01413208753539432 2.666955358959055 0.007654183800704909 5000#结局文件
pos_name chromosome position effect_allele reference_allele beta se z pval n_iids
rs11694978 2 102000210 A G 0.1642473177691523 0.0139500739683031 11.77393884379034 5.318212083690726e-32 5000
rs62156072 2 102000790 A G -0.010564912253069 0.014141346347707385 -0.7470938051652911 0.45500693932018343 5000
rs111408528 2 102001310 T C -0.010564912253069 0.014141346347707385 -0.7470938051652911 0.45500693932018343 5000
rs182612067 2 102001874 T C 0.07833004015877029 0.014098683660602683 5.555840675938812 2.7627879213433313e-08 5000
两个文件的表头都比较容易理解,但是还是附上介绍:
-
pos_name: SNP 的标识符(通常为 rsID),用于与基因型文件(.bim)中的变异一一对应。 -
chromosome: SNP所在的染色体编号(1–22、X、Y等)。 -
position: SNP在该染色体上的碱基坐标(以参考基因组版本为准,如GRCh37/38)。 -
effect_allele: 在这次关联分析中被视为“效应等位基因”(也即beta对应的等位基因)。 -
reference_allele: 与效应等位基因相对的另一等位基因。 -
beta: SNP对性状(exposure或outcome)的效应估计值。若性状是连续变量,则表示每增加一个效应等位基因,性状平均变化的单位数;若性状是二分类(如疾病状态),则多为对数优势比(log-odds)。 -
se: 对应beta的标准误。 -
z: z统计量,等于beta除以se,用于检验beta是否显著偏离0。 -
pval: 与z统计量对应的P值。 -
n_iids: 用于该SNP关联分析的样本量(number of individuals)。需要注意的是,MR-link-2默认需要各SNP至少有≥95%的最大样本量可用。
另外,需要PLINK基因型文件用于估算研究区段内SNP之间的连锁不平衡矩阵,逻辑上可以直接使用。当然,最好使用与分析的GWAS汇总数据统计人群结构(如欧洲、东亚等)一致的参考面板。
示例数据下载链接如下:
-
https://github.com/adriaan-vd-graaf/mrlink2/tree/main/example_files
02.运行示例
①基于下述代码执行MR-link-2分析:
python3 mr_link_2_standalone.py \--reference_bed example_files/reference_cohort \--sumstats_exposure sumstats_exposure.txt \--sumstats_outcome sumstats_outcome.txt \--out result_causal.txt
②需要注意的是,只要给MR-link-2完整的GWAS汇总统计,它就会自动定位并分析所有显著关联的区段。示例数据只输入了部分输入的结果,所以只有一个显著的结果:
cat result_causal.txt
#region var_explained m_snps_overlap alpha se(alpha) p(alpha) sigma_y se(sigma_y) p(sigma_y) sigma_x function_time
#2:101532661-103480976 0.99 1131 0.4193872544798938 0.0565878953774011 1.25110890260147e-13 0.1549295595449322 0.00624199019634178 5.3812527742659674e-136 0.564994403645872 0.09744000434875488
以上结果表明,在chr2:101532661–103480976区段,MR‑link‑2检测到显著的因果效应,并且该区段存在明显的水平多效性。
-
region:分析的基因组区段(染色体:起始–终止)。 -
var_explained:在LD矩阵中保留的方差比例,用于控制模型稳定性(此处 99%)。 -
m_snps_overlap:区段内用于估计的SNP数量(1131 个)。 -
alpha:因果效应估计值(暴露变量每增加1单位,结果变量增加0.419)。 -
se(alpha):因果效应的标准误(0.0566)。 -
p(alpha):因果效应显著性P值(1.25×10⁻¹³),小于0.05表明显著。 -
sigma_y:估计的多效性方差(反映SNP对结果的非暴露路径影响,≈0.155)。 -
se(sigma_y):多效性方差的标准误(0.00624)。 -
p(sigma_y):多效性存在的显著性P值(5.38×10⁻¹³⁶),表明水平多效性显著。 -
sigma_x:该区段对暴露性状的局部遗传度估计(≈0.565)。 -
function_time:本次估计耗时(≈0.097 秒)。
都到第二个版本了
看着确实不难运行
各位佬哥佬姐可以分析试试
这不点击关注一下