AlphaFold v3.0.1 conda版本详细安装与使用
2024年11月11日,AlphaFold 3.0.0源代码正式对外开源,仅限非商业用途使用,提供docker版本的使用介绍。
2024年12月底更新到了AlphaFold 3.0.1,解决了一些bug并提高了运行效率。
本文提供AlphaFold 3.0.1 conda版本的安装及使用方法。
本例安装环境:Ubuntu 22.04, RTX3060,64G内存,6T机械硬盘。CUDA runtime版本12.8。(nvcc -V 查看)。
目录
(1)下载源代码
(2)申请下载模型参数文件
(3)下载数据库
(4)配置alphafold3 conda环境
(5)安装alphafold3
(6)测试
参考资料

(1)下载源代码
git clone https://github.com/google-deepmind/alphafold3.git(2)申请下载模型参数文件
https://forms.gle/svvpY4u2jsHEwWYS6![]() https://forms.gle/svvpY4u2jsHEwWYS6
https://forms.gle/svvpY4u2jsHEwWYS6
新建文件夹models,将文件保存其中。
(3)下载数据库
需要的数据库如下:
- BFD (modified), by Steinegger M. and Söding J., modified by Google DeepMind, available under a Creative Commons Attribution 4.0 International License. See the Methods section of the AlphaFold proteome paper for details.
- PDB (unmodified), by H.M. Berman et al., available free of all copyright restrictions and made fully and freely available for both non-commercial and commercial use under CC0 1.0 Universal (CC0 1.0) Public Domain Dedication.
- MGnify: v2022_05 (unmodified), by Mitchell AL et al., available free of all copyright restrictions and made fully and freely available for both non-commercial and commercial use under CC0 1.0 Universal (CC0 1.0) Public Domain Dedication.
- UniProt: 2021_04 (unmodified), by The UniProt Consortium, available under a Creative Commons Attribution 4.0 International License.
- UniRef90: 2022_05 (unmodified) by The UniProt Consortium, available under a Creative Commons Attribution 4.0 International License.
- NT: 2023_02_23 (modified) See the Supplementary Information of the AlphaFold 3 paper for details.
- RFam: 14_4 (modified), by I. Kalvari et al., available free of all copyright restrictions and made fully and freely available for both non-commercial and commercial use under CC0 1.0 Universal (CC0 1.0) Public Domain Dedication. See the Supplementary Information of the AlphaFold 3 paper for details.
- RNACentral: 21_0 (modified), by The RNAcentral Consortium available free of all copyright restrictions and made fully and freely available for both non-commercial and commercial use under CC0 1.0 Universal (CC0 1.0) Public Domain Dedication. See the Supplementary Information of the AlphaFold 3 paper for details.
9个数据库中的前6个,可以用AF2.3中的数据库替代,建立软连接即可。
数据库保存在文件夹public_databases中。信息如下:
| 数据库 | 大小 | 描述 | AF3中的文件名 |
| BFD small | ~270 GB | BFD 是通过对 25 亿个蛋白质序列聚类而创建的 | bfd-first_non_consensus_sequences.fasta |
| MGnify | ~128G | 超过 24 亿个从宏基因组组装预测的非冗余序列 | mgy_clusters_2022_05.fa |
| PDB (mmCIF) | ~280G | PDB 收集了蛋白结构的实验数据 | mmcif_files(pdb_2022_09_28_mmcif_files.tar解压得到) |
| PDB seqres | 232.9M | PDB序列信息,fasta文件 | pdb_seqres_2022_09_28.fasta |
| UniProt | ~122G | uniprot存储蛋白的序列、功能等多种注释信息 | uniprot_all_2021_04.fa |
| UniRef90 | ~84G | UniRef 提供来自 UniProtKB 的序列聚类 | uniref90_2022_05.fa |
| NT | 81G | NCBI核苷酸序列库 | nt_rna_2023_02_23_clust_seq_id_90_cov_80_rep_seq.fasta |
| RFam | 228.4M | Rfam:RNA 家族的集合 | rfam_14_9_clust_seq_id_90_cov_80_rep_seq.fasta |
| RNACentral | 13.9G | RNAcentral:ncRNA序列数据库 | rnacentral_active_seq_id_90_cov_80_linclust.fasta |
后3个需要下载,修改fetch_databases.sh相应内容,下载即可。
cd alphafold3 && mkdir public_databasesbash ./fetch_databases.sh public_databases(4)配置alphafold3 conda环境
根据docker/Dockerfile安装文件,确认使用的Python版本为3.11和HMMER版本为3.4,创建一个新的Conda环境,并激活它:
conda create -n AF3 python=3.11
conda activate AF3编译安装hmmer 3.4,用于蛋白质序列搜索和比对:
mkdir hmmer_build hmmer
wget http://eddylab.org/software/hmmer/hmmer-3.4.tar.gz --directory-prefix ./hmmer_build
cd hmmer_build
tar zxf ./hmmer_build/hmmer-3.4.tar.gz
cd hmmer-3.4
./configure --prefix ../../hmmer
make -j8
make install
cd easel && make install
将安装位置加入PATH:
export PATH=path/to/hmmer/bin/:$PATH使用conda安装支持的GCC版本:
conda install gxx_linux-64 gxx_impl_linux-64 gcc_linux-64 gcc_impl_linux-64=12.4.0 -c conda-forge使用pip安装其他依赖项。首先,安装可以通过pip自动安装的包:
pip install -r dev-requirements.txt对于pip安装失败的包,使用pip补充安装。
pip install pint pydantic msgpack qcelemental optking matplotlib edgembar joblib ndfes运行:
(5)安装alphafold3
pip install . --no-deps --verbose需要的话,补充安装:
conda install catch2 zlib -c conda-forge(6)测试
{
"name": "2PV7",
"sequences": [
{
"protein": {
"id": ["A", "B"],
"sequence": "GMRESYANENQFGFKTINSDIHKIVIVGGYGKLGGLFARYLRASGYPISILDREDWAVAESILANADVVIVSVPINLTLETIERLKPYLTENMLLADLTSVKREPLAKMLEVHTGAVLGLHPMFGADIASMAKQVVVRCDGRFPERYEWLLEQIQIWGAKIYQTNATEHDHNMTYIQALRHFSTFANGLHLSKQPINLANLLALSSPIYRLELAMIGRLFAQDAELYADIIMDKSENLAVIETLKQTYDEALTFFENNDRQGFIDAFHKVRDWFGDYSEQFLKESRQLLQQANDLKQG"
}
}
],
"modelSeeds": [1]
"dialect": "alphafold3",
"version": 1
}以上保存为 fold_input.json
调整相应路径,运行:
python run_alphafold.py \
--json_path=./test_exmaples/fold_input.json \
--model_dir=./models \
--db_dir=./public_databases \
--output_dir=./af3_output \
--jackhmmer_binary_path=./hmmer/bin/jackhmmer \
--hmmalign_binary_path=./hmmer/bin/hmmalign \
--hmmbuild_binary_path=./hmmer/bin/hmmbuild \
--hmmsearch_binary_path=./hmmer/bin/hmmsearch \
--nhmmer_binary_path=./hmmer/bin/nhmmer
运行时长约4 h。
结果目录如下:
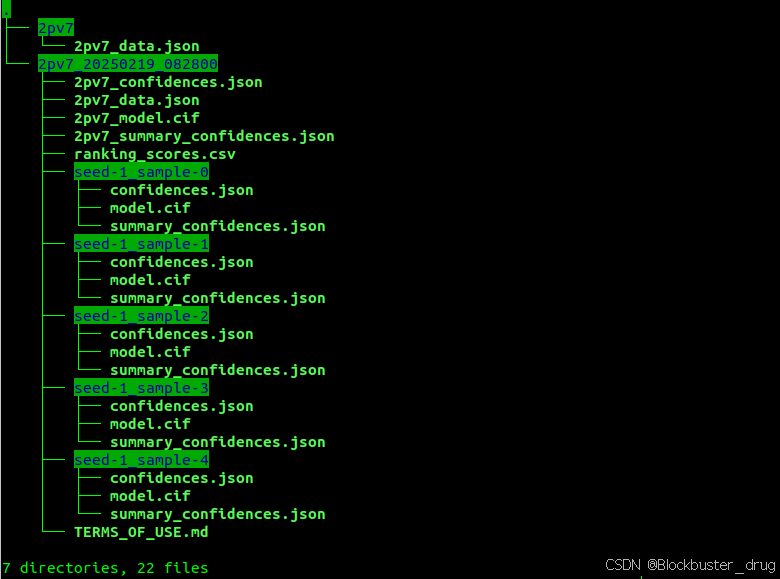
生成的测试输出文件主要包括:
1.2pv7_model.cif:这是排名第一的预测结果,以mmCIF格式保存,包含了蛋白质的三维结构信息,可以用PyMol打开,结构如下:

2.2pv7_confidences.json:包含排名第一的预测结果的置信度信息,以JSON格式提供,包含置信度信息,如 pLDDT(残基水平的置信度)、ipTM(界面预测置信度)和 PAE(预测对齐误差)等:
-
pLDDT(Per-Residue Confidence Score):表示每个残基的预测置信度,范围为0到100。pLDDT 越高,预测的可靠性越高。
-
pTM(predicted TM-score),来自模板建模(TM)得分。这衡量了整个结构的准确性。pTM > 0.5 意味着预测的复合物整体折叠结构可能与真实结构相似。
-
ipTM(Interface Template Modeling Score):用于评估蛋白质-蛋白质或蛋白质-配体界面的预测准确性。ipTM>0.8 表示高质量预测,<0.6则表示预测失败,0.6-0.8属于灰色区域,预测可能正确也可能不正确。
-
PAE(Predicted Alignment Error):以热图形式展示残基之间的预测误差。颜色越深表示预测越可靠。
-
ranking_score:计算公式为 0.8ipTM + 0.2pTM + 0.5disorder - 100has_clash。对整个复合体进行排序,使用ranking_score(越高越好)。该分数使用整体结构可信度(pTM 和 ipTM),但也包括惩罚冲突(clashes)和奖励不出现伪螺旋的无序区域,这些额外项意味着该分数只能用于结构排序。如果对特定的实体或相互作用感兴趣,例如在药物设计的场景下,可能需要通过特异于该链或链对的指标进行排序,而不是整个复合体。在这种情况下,请使用下文所述的每链或每链对置信度指标进行排序,例如chain_pair_iptm,chain_iptm,chain_ptm等等,参考AlphaFold3中文使用说明-CSDN博客
3.2pv7_summary_confidences.json:提供排名第一的预测结果的置信度摘要,同样以JSON格式呈现。
4.ranking_scores.csv:列出所有预测结果的排名分数。
(7)实际案例,运行结果与AF2.3比较
参考: AlphaFold3中文使用说明-CSDN博客
首先 编写输入信息的 json文件。
json文件主结构简要介绍如下:
{
"name": "Job name", #任务名称,被用于命名输出文件。
"modelSeeds": [1, 2], #随机种子整数列表,提供n个(n>=1)种子,获得n个预测结构。
"sequences": [
{"protein": {...}},
{"rna": {...}},
{"dna": {...}},
{"ligand": {...}}
], #序列字典的列表,每个字典定义一个分子实体。
"bondedAtomPairs": [...], #(可选)共价键合原子的列表。
"userCCD": "...", #(可选)用户提供的CCD。适用于:SMILES不足;自定义分子需要与其他实体结合;RDKit无法生成构象。
"dialect": "alphafold3", #输入JSON的风格,必须设置为alphafold3。参考JSON兼容性。
"version": 1 #输入JSON的版本,必须设置为1。
}
蛋白结构部分如下:
{
"protein": {
"id": "A", #一或多个大写字母,指定该蛋白链每个拷贝的唯一 ID。将被用于输出 mmCIF 文件。指定 ID 列表(如[“A”、“B”、“C”])意味着同源链有多个拷贝。
"sequence": "PVLSCGEWQL", #氨基酸序列
"modifications": [
{"ptmType": "HY3", "ptmPosition": 1},
{"ptmType": "P1L", "ptmPosition": 5}
], #(可选)翻译后修饰PTM列表,每个修饰都使用 CCD 和 1-based 的残基位置来指定。
"unpairedMsa": ..., #(可选)该链的多序列比对,使用 A3M 格式(等同于FASTA,但允许用"-"表示gap)指定。
"pairedMsa": ..., #(不推荐使用)
"templates": [...] #(可选)结构模板列表。
}
}
核酸,辅因子,配体结构json文件写法请参考:AlphaFold3中文使用说明-CSDN博客
fold_input_8I55.json
{
"name": "8I55",
"sequences": [
{
"protein": {
"id": ["A", "B"],
"sequence": "MEALVLVGHGSRLPYSKELLVKLAEKVKERNLFPIVEIGLMEFSEPTIPQAVKKAIEQGAKRIIVVPVFLAHGIHTTRDIPRLLGLIEDNHEHHHEHSHHHHHHHHHEHEKLEIPEDVEIIYREPIGADDRIVDIIIDRAFGR"
}
}
],
"modelSeeds": [3],
"dialect": "alphafold3",
"version": 1
}
与AF2.3的效果对比,后续再分享。。。
参考资料:
(1)Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630(8016):493-500. doi:10.1038/s41586-024-07487-w
(2)https://github.com/google-deepmind/alphafold3
(3)教程|使用Conda安装AlphaFold3-个人记录以及遇到的问题-CSDN博客