迈向生物界范围的基因表达分析-转录组综述-文献精读132
Toward kingdom-wide analyses of gene expression
迈向生物界范围的基因表达分析

亮点
-
目前已有超过30万个RNA测序(RNA-seq)实验公开可用,涵盖了大量藻类和陆地植物。
-
通过RNA-seq数据生成基因表达矩阵,现在可以在现代办公计算机上在几周内完成整个植物王国的处理。
-
来自不同物种的基因表达信息整合代表了一个尚未开发的资源,可以为植物生物学提供大量深刻见解。
-
全王国转录组分析不仅在阐明基因功能、发现新的生物途径和理解植物进化方面具有强大作用,还可以解决与单一物种分析相关的问题和局限性。
摘要
原始色素体生物(Archaeplastida)的基因表达数据正在呈指数增长,已有超过30万个RNA-seq实验可供使用,涵盖数百个物种。这些基因表达数据来自成千上万的实验,记录了不同器官、组织、细胞类型、(非)生物扰动和基因型中的基因表达。软件工具的进展使得在现代办公计算机上处理这些数据成为可能,并且可以在几周内完成,这使我们首次能够以全王国的方式研究基因表达。本文讨论了如何访问和处理这些表达数据,并概述了如何利用跨物种分析生成强大且稳健的基因功能与进化假设。
转录组学:理解基因功能的关键工具
一个生物体的信息内容编码在其DNA中,并通过转录过程表达为RNA。转录组(transcriptome)由所有信使RNA(mRNA)分子和非编码RNA分子组成,其中mRNA被翻译为蛋白质,而非编码RNA则在调控中发挥作用。可以使用多种方法对每个基因的表达进行定量分析,如Northern印迹杂交、qPCR、基因芯片、基因表达序列分析(SAGE)以及RNA测序(RNA-seq)(更多信息见参考文献[1])。 转录组提供了细胞内某一特定时间点的转录本快照,记录了生物体在不同器官、发育阶段、处理方式和条件下的基因表达情况。由于基因必须在正确的时间和空间中表达才能发挥功能,因此基因表达对理解基因功能(参见术语表)和发现生物通路至关重要,最终可帮助我们更好地理解植物王国的进化并改良作物。
随着新型转录组技术在灵敏度和成本效益方面的迅速发展,可公开获取的基因表达数据呈指数增长趋势 [2]。由Illumina、454、IonTorrent、Complete Genomics、Pacific Biosciences(PacBio)和Oxford Nanopore(ONT)等测序技术生成的原始数据被提交至主要的核苷酸序列数据库,即序列读取档案库(SRA)。截至2021年12月,绿色植物界(Viridiplantae)中已有超过30万个RNA-seq实验数据可供公开获取,涵盖数百个物种。尽管这些数据最初是为回答各种不同的研究问题而生成的,但我们可以将它们“再利用”,通过整合到跨大系统甚至整个植物王国的分析中,获取关于基因功能的新见解。
本综述讨论了这些数据如何被访问、处理并用于多种形式的比较转录组分析,以研究基因功能。我们展望,跨物种、全王国范围的分析将开辟更稳健、准确和全面的研究新途径,为基因功能预测与植物进化提供强有力的见解(见图1,关键图)。
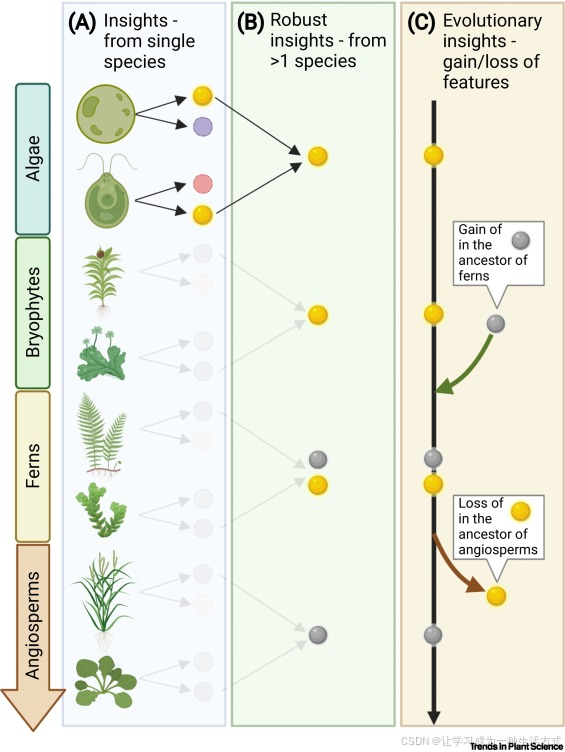
图1. 关键图。比较转录组学可以提供稳健的见解,并帮助我们从基因表达的角度理解进化。
转录组数据的分析可以生成以下方面的见解:(A)器官特异性基因表达(单一物种)、(B)保守性(多个物种)以及(C)基因进化(来自不同谱系的物种)。某些见解在多个物种中会被观察到(例如,“阿拉伯芥中的基因A在根部表达,水稻中的同源基因也如此”)。我们将这些称为(B)“稳健见解”,因为它们在多个物种中被独立观察到。 (C)“进化见解”则源自在特征的系统发育树中观察到的模式。例如,某些特征在藻类、苔藓和蕨类植物中可见,但在开花植物中却不可见(用金色圆圈表示),这表明这些见解背后的基因和转录程序在开花植物的祖先中已经丧失。相反,某些见解只在特定植物谱系(如由灰色圆圈表示的维管植物)中观察到,这表明这些见解背后的转录和基因组程序可能是突然出现的,或者是祖先基因的基因网络重编程。
植物中转录组数据的可用性和类型
美国国家生物技术信息中心(NCBI)的序列读取档案库(SRA)是一个公共数据库,属于国际核苷酸序列数据库合作组织的一部分 [3],该组织还包括欧洲生物信息研究所(EBI)和日本DNA数据银行(DDBJ)。提交到这三个组织的数据在三个数据库中进行镜像,并可供研究社区使用。由于大多数期刊要求在研究论文接受后公开发布测序数据,因此这些公共数据库不断增长,包含新的基因表达数据。
截至2022年4月,SRA已有超过60,000太字节的开放访问基因序列读取数据,来自不同的测序技术和样本类型(即DNA或RNA测序)。对于真核生物,SRA中共有8,054,255个实验,其中77%(6,196,620个)属于后生动物,17%(1,354,735个)属于绿色植物(绿色植物界),其余6%属于其他真核生物。在可用的绿色植物SRA数据中,23%(315,701个)来自基因表达研究(此后称为RNA-seq;见图2A)。尽管大多数数据来自Illumina短读测序,但来自新测序平台(如BGISEQ、PacBio单分子实时(SMRT)测序和ONT)的RNA-seq数据在过去4年中快速增加(见图2A)。BGISEQ正在成为Illumina在植物转录组学分析中的替代选择 [4,5],而长读测序(如PacBio SMRT和ONT)是克服短读测序技术局限性的非常有吸引力且合适的选择。例如,尽管与短读测序相比,PacBio和ONT测序更昂贵且更容易出错,但它们能够揭示全长转录本,从而识别转录本异构体 [6]。此外,ONT是目前唯一可以直接对RNA进行测序而不需要反转录或扩增的商业化方法,这使得RNA修饰的识别成为可能 [7, 8, 9]。有关不同测序技术比较的更多详细信息,请参考相关综述 [4,10,11,12]。
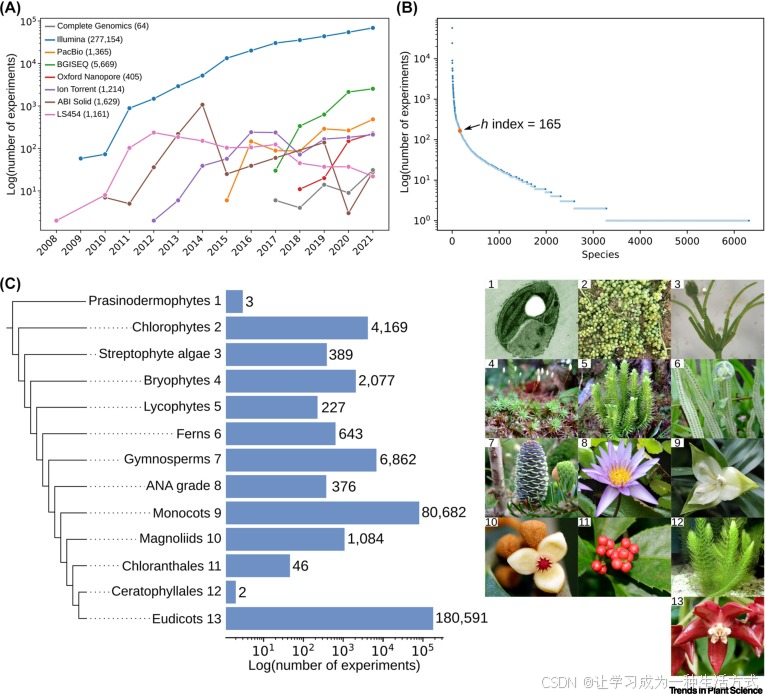
图2. 绿色植物界公开可用的RNA测序(RNA-seq)数据
(A) 来自不同测序平台的RNA-seq数据数量。y轴表示从2008年到2021年(x轴)每年提交到国家生物技术信息中心序列读取档案库(SRA)的样本数量(以对数比例表示)。 (B) 绿色植物的转录组h指数。y轴显示每个物种的实验数量(以对数比例表示)(x轴)。物种按照实验数量从高到低排序,并用数字表示。h指数计算为至少有h个物种,每个物种至少有h个RNA-seq实验,h指数以橙色显示并用箭头突出。 (C) 提供Illumina RNA-seq数据的绿色植物主要谱系(x轴为对数比例)。条形图右侧的数字表示数值。谱系名称旁的数字与右侧图片的编号对应。图片1和3来自MicrobeWiki,12来自Wikipedia,2和4-8来自B.C. Ho,9-11和13来自L.M.J. Chen。
绿色植物的RNA-seq数据在分类上分布不均。由于Illumina是生产大多数RNA-seq数据的平台,占比96%(266,265),它为比较转录组学提供了独特的机会。然而,约50万个绿色植物物种中,只有很小一部分(1.3%;6,496个物种)拥有RNA-seq数据[13]。尽管计算得到的转录组h指数很高(165;图2B),大多数数据来自双子叶植物(65%)和单子叶植物(29%)(图2C,蓝色条形)。其他谱系,如裸子植物、绿藻和苔藓植物,仅占总RNA-seq数据的1.6%,而其他类别的物种则不到1%。这些分类差距限制了比较转录组学研究仅限于少数植物群体,通常是那些具有经济重要性和作为模式物种的植物,忽视了绿色植物所包含的巨大信息多样性。目前可用的信息(或缺乏的信息)突显了研究这些代表性较少的群体的必要性,包括链格孢藻、绿藻、角果类、绿色海藻门、石松植物以及ANA级别的被子植物。高质量参考基因组和基因注释的可用性将使我们能够从基因表达的角度,深入了解绿色植物的重大进化过渡。数据集的质量至关重要,以确保得出的结论不是由基因组不完整或基因注释不准确等人为因素造成的。随着基于去新转录组组装的RNA-seq分析逐渐流行,这一问题尤为突出,因为这种方法成本较低,但与基因组组装面临相同的挑战,且具有冗余性。最近的生物信息学方法已能在高端工作站上自动处理超过10,000个RNA-seq样本/天,使得所有植物数据的处理时间不超过1个月[14,15]。
尽管有大量RNA-seq数据可用,但缺失或不一致的元数据(例如采样的组织)限制了数据在比较转录组学中的应用价值。元数据通常可以在NCBI的RunInfo表格中找到,并以标准表格格式下载。然而,某些列(如BioSample、SampleType)的内容可能会有歧义,因为它们根据提交情况包含不同的信息。例如,研究人员在样本类型的命名中使用不同的名称来表示相同的器官(如叶子和叶片),这被指出是比较转录组学中的主要问题之一[16,17]。当某些列为空且无法检索所需信息时,这一问题会加剧。尽管有一些自然语言处理方法可以自动提取器官类型[14],但大多数样本的自动注释仍然是一个挑战。因此,未来解决这一问题的挑战之一是寻找能够规范研究人员在实验中提供信息的系统,特别是对于样本类型。
全王国范围的比较转录组学
比较转录组学是一个快速发展的领域,旨在理解同一物种不同样本之间或不同物种之间转录组的变化。类似于旨在识别基因组中保守基因和调控区域的比较序列分析,比较转录组学可以识别保守的基因表达模式,从而找到在不同生物体中执行相同功能的同源基因,突出对生物过程至关重要的基因,并从基因表达的角度研究基因的进化。各种比较转录组分析包括基因表达差异(DGE)、基因表达图谱、共表达网络和表达数量性状基因座(eQTL)分析,下面我们将讨论这些分析(见图3)。
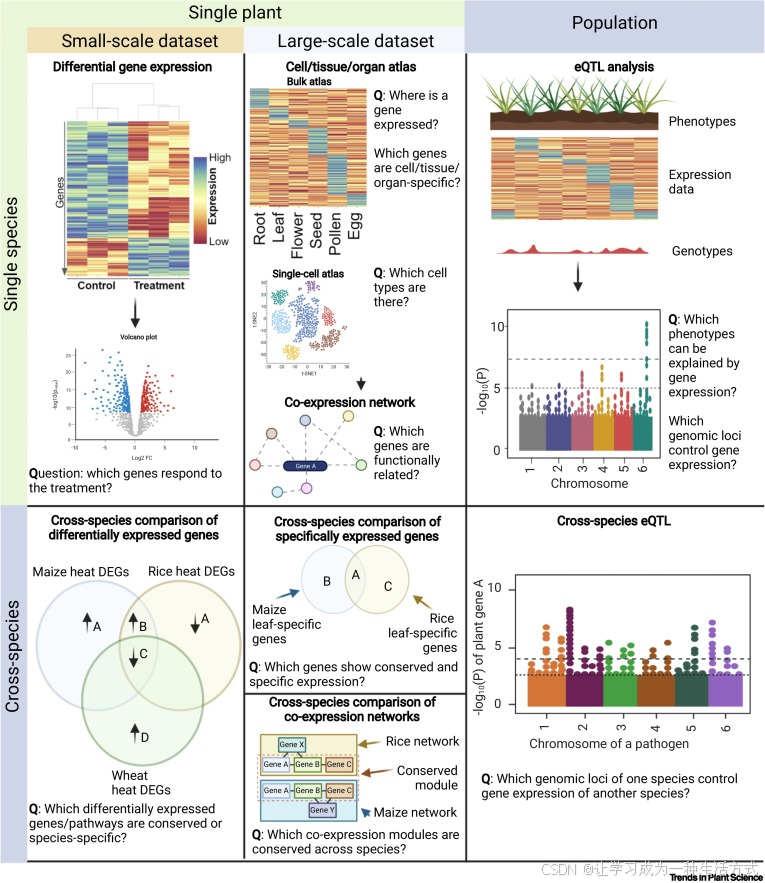
图3. 单物种与跨物种比较转录组学方法的不同形式
这些方法分为单物种分析(绿色行,顶部)和跨物种分析(蓝色行,底部)。分析可以进一步分为小规模分析(例如,差异基因表达,左列)、大规模分析(表达图谱,共表达网络分析,中列)和种群数据集分析(表达数量性状基因座(eQTL),右列)。每种分析都能回答特定的问题。此图由BioRender制作。
跨物种差异基因表达(DGE)
单物种的DGE分析旨在通过识别在施加扰动时基因表达发生变化的基因,揭示那些对遗传或环境扰动(例如,干旱中的生存)至关重要的基因(图3,顶部行,左列)。已经开发了多种执行DGE分析的工具[18],结合富集分析后,可以揭示扰动所影响的基因功能和生物过程[19]。
跨物种的DGE分析提出了更多关于植物如何响应扰动的问题。植物的响应是否在同一属、同一家族甚至整个植物王国中是保守的?响应的哪些部分是物种特异性的?哪些基因对于响应特定扰动至关重要?尽管这些问题可以通过单一物种的DGE分析来识别,如果同源基因在多个物种中表现出差异表达,跨物种分析可以增加对观察结果的信心,尤其是在处理噪声较大的生物学数据时(框1)。此外,在多个物种中发现的具有未知功能的保守差异表达基因(DEGs)可能会成为进一步分子表征的有趣目标[20]。
框1:转录组学的警告
选择合适的细胞/组织/器官和采样时间会极大地影响基因表达,从而影响我们的推论。例如,转录组学通常用于研究压力反应,叶片通常是首选样本。因此,大多数压力相关的基因表达来自叶片,但其他器官可能对压力的反应不同[98]。此外,时间因素使得生物学过程的研究变得复杂[99]。例如,年龄可以显著改变表达模式。一项关于葡萄的研究发现,器官的转录组根据年龄进行聚类,而非根据器官类型(例如,老叶和老根比年轻叶和老叶更相似[100])。此外,约50%的绿藻和约30%的陆生植物基因呈昼夜变化[50],这表明我们的转录组学推论可能会随昼夜循环参数和采样时间的不同而发生变化。对处理的反应也高度依赖于时间,因为对刺激的转录组反应可能会从几分钟到几天发生变化[101,102]。此外,基因表达与压力反应的关系难以解读;例如,DEGs不一定在压力反应中起重要作用,而可能是压力反应的结果。因此,进一步的实验是必要的,以验证DEGs的参与及其在缓解压力中的作用。转录组学分析的其他局限性包括mRNA与蛋白质丰度之间的可变相关性,这阻碍了基因型与表型之间的联系[103]。除了这些局限性,批次效应是未考虑时可能影响结果解释的主要源之一[104]。因此,由于这些和其他变量以及高昂的实验成本(一个简单的处理/对照实验可能轻松超过1000美元),目前的研究通常只能捕捉到转录组反应的一个小部分。
为了说明一个常被忽视的参数(基因型)如何改变转录组学分析的结果,我们展示了两种耐盐基因型在水稻中的下调和上调基因集(图I)。令人惊讶的是,这两种基因型在DEGs的表现上几乎没有一致性。共表达网络的分析可以捕捉转录组学中的另一个变化例子。ATTED-II数据库比较了基于微阵列和RNA-seq数据构建的共表达网络的可重复性[105]。尽管这些网络在预测基因功能方面表现相同,但它们在共享的边数上只有 modest 的重合,阿拉伯芥、稻米和玉米的Jaccard指数分别为0.136、0.041和0.055。
总结
总之,有许多技术性和生物学上不完全理解的原因导致我们在基于转录组数据的推论中看到较弱的一致性。因此,“缺乏证据并不等于证据的缺失”这一格言在转录组学中非常真实。此外,与其将精力集中在少数模式植物上,这些差异强调了需要通过比较转录组学迈向更稳健的推论。
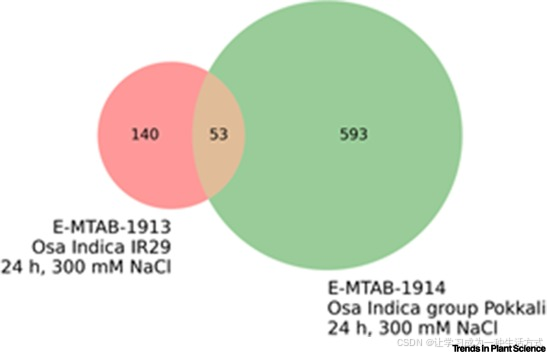
图I. 水稻两种耐盐品种中DEG反应的相似性
数字表示两个实验中上调和下调基因的数量。此数据集的Jaccard指数为 53 / (140 + 53 + 593) = 0.067。
在跨物种DGE分析的研究中,初步步骤与单物种分析(图4A)的方法类似,但额外的重点是识别同源基因,以便可以在物种之间进行比较。尽管跨物种映射可用于关系密切的物种[21],但这里讨论的大多数研究和文献中发现的研究依赖于比较同源基因。准确的同源性识别是比较转录组学分析的第一步,近年来已开发了不同的工具,包括OrthoFinder [22]、InParanoid [23] 和SonicParanoid [24](更多细节请参见[25])。
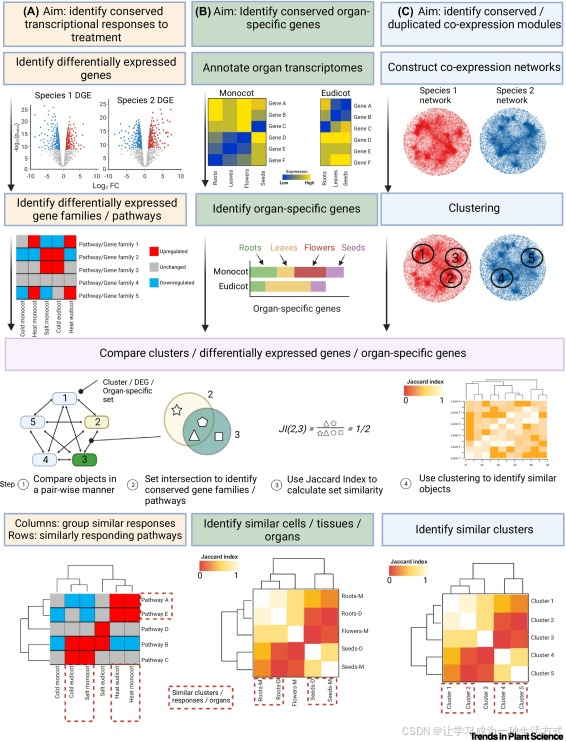
图4. 从不同处理、器官和簇的基因表达角度进行的比较转录组学分析示例
这三种分析分别考察了:(A) 对处理的保守转录反应(黄色,左列),(B) 器官特异性基因(绿色,中列),和(C) 保守/重复的共表达模块(蓝色,右列)。 (A) 对处理的保守转录反应的识别从两个物种的差异基因表达(DGE)分析开始,接着识别对两个物种都常见的上调和下调基因家族/通路。 (B) 对保守器官特异性基因的识别从基因表达数据的注释开始,识别两个物种(即单子叶植物和双子叶植物)中的器官特异性基因。 (C) 对保守或重复的共表达模块的识别从两个物种的共表达网络开始,经过聚类分析以分离出功能相关的基因组。对不同对象(分别为步骤(A)、(B)和(C)中识别的差异表达基因、器官特异性基因和簇)进行比较,通常包括按对进行的所有对象的比较(步骤1),计算它们的相似性,例如通过Jaccard指数(步骤2和3)。然后可以通过聚类(步骤4)来识别相似的对象。这些分析的结果可以通过簇图可视化,显示不同对象之间的关系。此图由BioRender.com制作。
接下来,比较分析可以根据研究兴趣以多种方式进行。通常,它将涉及按基因家族和/或生物过程层面对差异表达基因(DEGs)进行对比(图4,左列),比较通过集合操作(如集合交集,用于识别两个物种中响应相似的基因家族/生物过程,或集合差异,用于识别物种特异性反应)进行。为了可视化多个物种在各种条件下的趋势,可以使用多重对比集来执行显著富集的生物过程或基因家族的层次聚类[26,27](图4)。
跨物种的差异表达基因(DGE)分析已广泛用于理解作物与野生植物之间的差异[28,29],不同物种对非生物和生物压力的反应[30,31],发育过程中的差异[29,32,33],以及系统基因组学[34, 35, 36, 37]。例如,一项关于番茄的研究观察到商业品种在氮胁迫下次生代谢物生产的诱导更强,并确定了改善植物压力耐受性的潜在目标[28]。另一项研究比较了玉米和高粱在真菌病原体中的转录组反应,揭示了保守和物种特异性反应的混合模式[30]。
跨物种的DGE分析已显示出在响应非生物压力时的不同保守模式。例如,红枸杞(Lycium ruthenicum)和黑枸杞(Lycium barbarum)之间的比较使用转录组和代谢组数据,识别出多种可能对更具抗逆性的L. ruthenicum耐非生物压力至关重要的基因和代谢物[29]。此外,九个草本物种的比较研究显示,尽管所有植物在4°C适应后都能耐受低温,但适应的植物中观察到的差异表达基因在九个物种之间不同,表明冷适应在这些植物中是独立进化的[38]。最后,比较陆生植物拟南芥(Arabidopsis thaliana)、绿藻(Chlamydomonas reinhardtii)和蓝藻(Cyanophora paradoxa)在多种非生物压力下响应的差异表达基因(DEGs)揭示,这些高度分化的物种在参与光合作用和初级代谢的基因上显示出保守的下调,表明这种下调是全王国范围的压力响应程序[26]。
比较表达模式分析与图谱
通过细胞、组织、器官、发育阶段、处理或条件的基因表达模式可以用来阐明基因的功能,因为基因应该在其需要的时间和位置表达。基因表达图谱可以识别普遍表达的家族基因,这些基因可以作为qPCR中的标准化参考[39, 40, 41, 42]。此外,识别普遍表达基因的启动子可以用于在作物的多个组织/器官中驱动转基因表达[43]。然而,基因表达图谱也可以识别特定于某一器官表达的基因,从而揭示对器官形成和功能至关重要的基因。
跨物种比较基因表达图谱有助于我们理解不同谱系中生物过程的保守性和分化[44]。对于关系密切的物种,如属级物种或栽培植物及其野生亲缘种,比较同源器官是可行的[45, 46]。然而,对于远缘物种(例如开花植物和单细胞藻类),可能无法定义可比的样本,在这种情况下,采用的是全对全的比较方法[17],其中所有器官/样本的相似性被比较,无论它们是否具有同源性。比较器官特异性基因需要识别同源基因和/或同源组。前者方法在许多研究中使用,但仅能分析少数基因家族,而后者方法正受到越来越多的关注,但它包括可能在功能上发生分化的重复基因[25]。一些方法首先使用适当的度量识别器官特异性基因[47],然后通过基因家族的水平比较这些基因集[48, 49, 50](图4B)。采用这种方法,研究表明,具有保守、普遍表达的基因往往在更多物种中发现,并具有重要功能[48, 51]。
跨物种比较样本相似性的多种方法存在。相似的样本基于相似性度量(欧几里得距离和曼哈顿距离、皮尔逊和斯皮尔曼相关系数、Jaccard指数)进行分组,这些度量是通过器官特异性基因家族的集合计算得出的(更多细节请参见[52, 53])(图4)。然后,采用多种无监督和监督聚类方法来分析不同物种样本中的基因表达的一般模式。无监督聚类方法,如主成分分析、k均值聚类和层次聚类,可用于指示哪些样本是相似的[21, 49, 50, 54]。监督聚类方法(例如,偏最小二乘判别分析(PLS-DA)和正交PLS-DA)可以进一步细化高度相似样本的分离,正如一项在六个甘蓝物种中的研究所示[49]。
多项研究表明,许多植物器官的基因表达模式似乎是保守的[17, 48, 49, 50]。值得注意的是,在维管植物的根[17, 54]和叶片[17, 55]、豆科植物的根瘤[56, 57]、被子植物的花粉[17]、以及拟南芥和药用毛果的顶端生长细胞[58]之间观察到了高度的保守性。水稻和拟南芥的根部单细胞解析揭示了保守的进化性和物种特异性的基因表达特征[59]。相反,玉米和拟南芥的顶端分生组织(SAM)的比较揭示了支配干细胞调控和身份的不同基因集[60]。然而,一项包含五个物种的最新研究显示,SAM的转录组是保守的[17]。类似地,尽管以前关于种子中基因表达的研究显示三种草本植物的种子特异性转录组之间缺乏保守性[61],但最近的一项研究却得出了相反的结论:银杏、水稻、玉米、西红柿和拟南芥的种子特异性转录组显著相似[17]。这些相反的结果突显了在进行比较转录组学时,进行更全面的分类学采样(以物种为单位)以得出稳健结论的重要性(见图1)。然而,在进行这些比较时,必须特别注意是否包含物种间的进化关系信息[62]。
表达图谱能够揭示基因的时空调控的新见解,这是比较基因组学分析无法实现的。例如,拟南芥[63]、甘蓝[49]和小麦[64]物种在胚胎发育过程中表现出典型的沙漏型表达模式,在形态学定型阶段,基因的表达变化最小,而在此之前和之后变化较大。然而,这种转录组沙漏型模式是否仅限于胚胎发生仍存在争议[65]。其他进化模式被提出,如拟南芥、稻米和大豆的生殖发育过程中出现的“进化膨胀”[66],以及玉米和高粱花序发育的反向沙漏模型[67]。另一项大规模研究表明,尽管跨越超过10亿年的大的系统发育距离,昼夜基因表达仍是保守的[50, 68]。此外,倍性植物的转录组图谱揭示了亚基因组的非对称表达谱,并且与来自二倍体个体的样本存在明显差异[49, 69, 70]。最后,对器官特异性基因的系统发育谱系分析显示,器官很可能是通过重新利用现有的遗传材料进化而来,而非通过招募新基因[17]。
跨物种比较共表达网络
共表达网络分析是一种强大的方法,利用基因表达数据将功能相关的基因联系起来。该分析基于这样一个观察结果:功能相关的基因往往在不同器官、组织和处理条件下具有相似的表达模式。过去十年中,关于共表达网络的构建[71, 72]、分析[72, 73]和在线资源[17, 72, 74]的许多方面已被讨论。通过比较共表达网络,可以识别跨物种保守的共表达基因群体(称为“保守模块”或“簇”)(回顾见[75])。此外,由于其独特且有用的特点,比较共表达分析揭示了基因功能和生物通路的进化。
首先,保守模块可以用于在不同生物之间转移功能注释。仅通过序列相似性方法识别真正的同源基因可能会很困难,因为植物中存在复杂的基因家族和重复模式[76, 77, 78]。具有多个同源基因的基因往往有一个具有保守网络邻域的同源基因,这表明它们是“真正的”同源基因[79]。因为比较共表达分析识别了具有相似表达模式的同源基因,所以识别保守模块可以更有信心地揭示“真正的”同源基因[80, 81],并允许在例如模式植物和作物之间更稳健地转移功能信息。
其次,保守模块可能代表一个真实的功能相关基因群体(即“稳健见解”)(图1)。这一说法基于这样一个假设:假阳性共表达关系(框1)不太可能出现在多个物种中[20]。因此,揭示保守模块及其中的基因可以用于去除假阳性并突出具有相关生物功能的基因[20, 82, 83]。
第三,比较共表达网络去除了基因表达数据必须来自同源器官的要求,因为基于网络的比较通常是基于相似的共表达簇或邻域,而不是基于相似的转录组条件[83]。
比较共表达网络分析可以从构建两个或更多物种的共表达网络开始。然后,对这些网络进行聚类,提出功能相关基因集以供比较(图4,右列)[20, 84]。为了揭示相似的簇,典型的分析计算两个簇共同包含的具有相似特征(例如,同源组、基因家族或Pfam域)的基因数量。常用的度量是Jaccard指数,其范围从1(两个簇包含相同的同源基因集)到0(两个簇完全不相似)(图4C)。簇的相似性也可以通过网络可视化,其中节点代表簇,边表示相似的簇之间的连接[74]。保守的共表达模块已在多个物种中观察到,并用于研究种子寿命[85]、光合作用[86, 87]和细胞壁发育[20, 88, 89]。
尽管通常跨物种比较簇,但该分析也可以比较同一物种内的簇,从而揭示重复簇(图4C)[90]。重复簇代表已重复的生物通路,并执行相关的生物功能[91]。例如,拟南芥在花粉发育过程中使用两种特殊的酚类通路[92],以及植物的其他部分[93]。这些通路已被复制并次级专业化,以在不同的组织中产生相关的代谢物。类似地,角质层和亚角质层是细胞壁中合成的脂质生物聚酯,由在空中组织和根中分别表达的重复基因模块合成[94]。其他重复模块在植物中也经常出现。例如,拟南芥的细胞壁合成模块至少已经重复三次,以支持初级、次级和顶端生长细胞壁的合成[90],并且有证据表明,苔藓P. patens也独立复制了其细胞壁模块[75]。如果仅使用基因组学方法,是无法检测到这些重复模块的,因此这展示了基于网络的分析的优势。在线工具,如CoNekt/Evorepro和protist.guruv数据库,允许在植物和藻类中检测保守和重复模块[17, 74]。
跨物种eQTL分析
QTL分析与全基因组关联研究(GWAS)结合,在阐明与表型变化相关的基因组特征方面提供了重要信息。eQTL研究旨在识别解释基因表达的基因组位点,而转录组全基因组关联研究(TWAS)旨在将基因表达与表型联系起来。尽管目前跨物种eQTL/TWAS的出版物不多,但近年来已有几种创造性的方法。
从独立的eQTL/GWAS中识别出的基因可以在物种之间进行比较。例如,659个高粱品种的GWAS分析识别出239个基因,推测这些基因驱动气孔导度性状的变异[95]。此外,其中77个基因是拟南芥基因的同源基因,涉及水分利用效率的各个方面,例如气孔开闭(24个基因)、气孔/表皮细胞发育(35个基因)、叶/血管发育(12个基因)或叶绿素代谢/光合作用(8个基因)[95]。这表明,气孔导度的遗传基础在一定程度上是保守的,并且跨物种的eQTL/GWAS分析可以用来识别这些保守的、高置信度的与关键生物特征相关的基因。
其他跨物种eQTL/TWAS方法为共生、寄生和致病性提供了独特的见解。这些跨物种的方法通常研究物种A的基因型如何影响物种B基因的表达。例如,为了研究一个物种中的遗传位点如何影响另一个相互作用物种的基因表达,一项eQTL分析研究了被98个基因型的二倍体寄生线虫 Meloidogyne hapla 感染的同源 Medicago truncatula 亲本的基因表达[96]。分析揭示了213个富含转录因子的Medicago基因,这些基因主要受三个寄生虫联会群体的控制,作者将这些基因称为宿主表达调节位点。在另一项研究中,使用了96个病原菌Botrytis cinerea基因型群体,同时识别控制病原菌和宿主拟南芥基因表达的eQTL[97]。识别的eQTL是明确的方向性,因为它们必须控制病原菌基因的表达,而病原菌基因必须控制宿主基因的表达。此外,该分析主要揭示了小效应的跨位点eQTL热点,没有识别出影响宿主或病原菌转录组的热点重叠。总体而言,这些结果表明,病原菌基因组对宿主和病原体转录调控的基础出乎意料地独立。
结语与未来展望
公开可用的基因表达数据是需要被提炼才能具有价值的新“石油”。超过30万个RNA-seq实验数据覆盖数百个绿色植物物种,提供了新的激动人心的机会,能够生成关于植物分子电路的稳健、可重复的推论,这些推论在实验和田间条件下都可能是有效的。跨不同系统发育层次的物种间推论比较将帮助我们进一步理解植物的陆生化、专业代谢、植物-微生物相互作用、非生物胁迫抗性和其他重要性状背后的基因和通路。然而,由于下载、注释、质量控制和数据分析的任务看似艰巨,目前大多数数据尚未被超越原始研究进行分析。然而,近期软件工具的进展使我们能够在不到1个月的时间内处理所有可用的植物物种数据。我们预见,在未来几年,比较转录组学分析将成为主流,通过机器学习方法处理数百万基因在数百个物种中的基因表达数据,以解释植物的分子电路和进化(见“突出问题”)。
突出问题(Outstanding questions)
-
当前可用的数据涵盖了数百万个植物基因的表达信息。一个大规模的比较转录组研究能够纳入多少类群?其推论在进化树上可以延伸多深?
-
从日益丰富的转录组数据中(如小RNA、长非编码RNA、可变剪接、RNA甲基化等)进行大规模比较分析,可以带来哪些新见解?
-
除了同源性/直系同源信息之外,从表达数据中推断出的哪些其他特征(例如GC含量、共表达网络结构)可以用于提取有价值的信息?
-
将会开发出哪些新工具,用以快速且准确地分析大规模的比较转录组数据?
-
面对海量数据,我们能否构建结合经典计算生物学与机器学习的方法,来预测基因功能、调控方式、亚细胞定位、蛋白质互作等?
-
相当大比例的RNA-seq样本缺乏注释,这使得我们难以据此构建有信息量的表达图谱。我们如何确保研究人员在提交数据到序列读取档案(SRA)时提供详尽的元数据和注释?
-
复杂的实验设计常常使得自动判断研究中哪些样本应当被比较变得困难。我们如何配置元数据,使其支持机器可读的实验设计解析?
-
尽管自然语言处理方法可以处理元数据,但目前尚不清楚应如何利用这些方法提取有价值的信息。