TWAS、GWAS、FUSION
全基因组关联研究(GWAS,Genome-Wide Association Study)是一种统计学方法,用于在全基因组水平上识别与特定性状或疾病相关的遗传变异。虽然GWAS可以识别与性状相关的遗传信号,但它并不直接揭示这些遗传变异如何影响生物学过程和机制。
转录组关联分析(TWAS,Transcriptome-Wide Association Study)是一种扩展的GWAS方法,它结合了基因表达数据(eQTL,expression Quantitative Trait Loci)来增强对遗传信号的理解。TWAS通过以下方式帮助从统计学信号转向生物学机制:
-
整合表达数据:TWAS不仅分析遗传变异,还分析这些变异如何影响基因的表达。通过比较不同条件下(如不同组织、不同环境或不同疾病状态)的基因表达模式,可以识别出可能参与性状调控的基因。
-
构建预测模型:利用统计模型和机器学习方法,TWAS可以构建基因表达的预测模型。这些模型能够预测未知样本中基因的表达水平,从而帮助理解遗传变异如何通过影响基因表达来影响性状。
-
映射到转录水平:TWAS直接将GWAS信号与基因转录水平的变化联系起来,从而提供了一种机制上的解释。这对于理解非编码区段的变异尤其重要,因为这些变异可能通过影响基因表达来影响性状。
-
精细定位:通过共定位分析(即在遗传图谱中寻找与多个性状相关的变异)和条件分析(考虑不同环境或生物学条件对性状的影响),TWAS可以进一步细化与性状相关的遗传变异的位置,从而更精确地定位潜在的致病基因。
-
功能挖掘:TWAS可以帮助识别那些在特定性状中起关键作用的基因,这些基因可能是新的药物靶点或生物学研究的焦点。
FUSION作为TWAS的一个经典工具,它通过以下方式帮助研究人员:
- 优先致病基因:FUSION可以帮助研究人员从大量候选基因中优先考虑那些最有可能与疾病相关的基因。
- 共定位分析:FUSION可以进行共定位分析,以识别与多个性状相关的遗传变异。
- 条件分析:FUSION允许研究人员探索不同条件下性状的遗传基础,从而提供更深入的生物学见解。
全基因组关联研究(GWAS)中识别的遗传变异大多位于基因调控区域,这些变异可能通过影响基因表达来影响复杂性状。然而,由于样本数量的限制和高成本的基因表达测量,大规模地测量基因表达具有一定的挑战性。因此,许多表达-性状的关联尚未被检测到,尤其是那些效应量较小的关联。为了解决这些问题,提出了替代方法,其中转录组全关联研究(TWAS)已成为一种常见且易于执行的方法,用于识别在未直接测量表达水平的个体中,与复杂性状显著相关的基因。
-
GWAS中识别的变异:在全基因组关联研究中,许多与性状相关的遗传变异被发现位于
基因的调控区域,而不是基因编码区域。这些调控区域的变异可能通过影响基因的表达来影响性状。 -
基因表达测量的挑战:由于样本数量的限制和测量成本较高,大规模地测量基因表达具有一定的难度。这限制了对表达-性状关联的检测,尤其是那些效应量较小的关联。
-
TWAS的提出:为了克服这些挑战,提出了转录组全关联研究(TWAS)作为一种替代方法。TWAS通过分析基因表达数据与性状之间的关联,来识别与复杂性状显著相关的基因。
-
TWAS的优势:TWAS已成为一种常见且易于执行的方法,它能够在未直接测量表达水平的个体中识别与复杂性状显著相关的基因。这种方法特别适用于检测那些效应量较小的表达-性状关联。
-
TWAS的应用:TWAS的应用可以帮助研究人员更好地理解遗传变异如何通过影响基因表达来影响复杂性状。这对于发现新的生物学机制、识别潜在的疾病相关基因以及开发新的治疗策略具有重要意义。
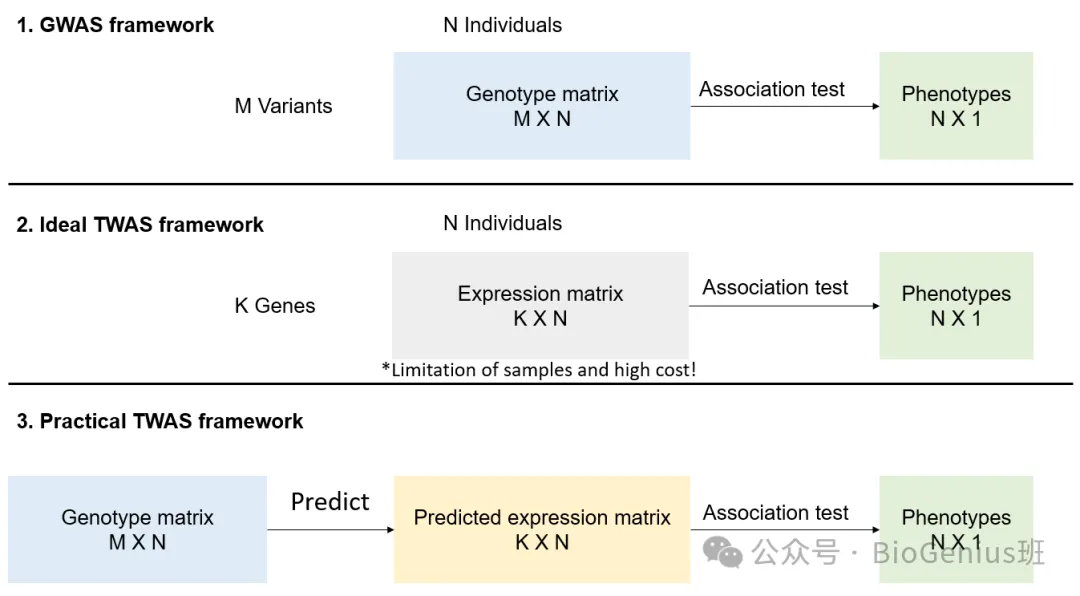
这张图展示了三种不同的基因组学研究框架:GWAS(全基因组关联研究)、理想中的TWAS(转录组全关联研究)和实际中的TWAS框架。
1. GWAS框架
- M Variants(M 变异):研究中考虑的遗传变异的数量。
- N Individuals(N 个体):参与研究的个体数量。
- Genotype matrix(基因型矩阵)M x N:一个包含所有个体所有遗传变异的矩阵。
- Association test(关联测试):用来检测基因变异与表型之间的统计关联。
- Phenotypes(N x 1 表型):每个个体的表型数据。
2. 理想TWAS框架
- K Genes(K 基因):研究中考虑的基因数量。
- Expression matrix(表达矩阵)K x N:一个包含所有个体所有基因表达水平的矩阵。
- 同样进行关联测试,以确定基因表达与表型之间的关联。
- 表型数据与GWAS框架相同。
这个理想框架的挑战在于需要大量的样本和高昂的成本来测量所有基因的表达水平。
3. 实际TWAS框架
- 从基因型矩阵开始,该矩阵包含遗传变异信息。
- Predict(预测):利用基因型信息来预测基因表达矩阵。
- Predicted expression matrix(预测表达矩阵)K x N:预测的基因表达水平矩阵。
- 然后使用关联测试来确定预测的基因表达与表型之间的关联。
- 输出与前两个框架相同,是每个个体的表型。
总结
- GWAS 关注于检测遗传变异与表型之间的关联。
- 理想中的TWAS 关注于检测基因表达与表型之间的关联,但成本较高。
- 实际中的TWAS 通过预测基因表达,以较低的成本实现与理想TWAS相似的目标。
实际TWAS框架通过预测基因表达水平,提供了一种成本效益更高的方法来研究基因表达与表型之间的关联。
3.TWAS分类
TWAS(Transcriptome-Wide Association Study,转录组全关联研究)是一种结合基因表达数据和遗传变异数据来识别与特定性状相关联的基因的方法。这种方法特别适用于研究那些可能通过调控基因表达来影响性状的遗传变异。TWAS 可以分为个体水平和汇总水平两种类型:
个体水平 TWAS
个体水平的 TWAS 使用每个个体的基因型和表型数据来进行基因表达的预测和关联测试。这种方法通常涉及以下步骤:
-
基因型数据:收集每个个体的基因型数据,这些数据通常来自于全基因组关联研究(GWAS)。
-
表型数据:收集每个个体的表型数据,这些数据可以是任何可测量的性状,如身高、疾病状态等。
-
表达预测:使用统计模型或机器学习方法,基于基因型数据预测每个个体的基因表达水平。
-
关联测试:将预测的基因表达水平与表型数据进行关联测试,以识别与性状显著相关的基因。
一个例子是 PrediXcan 方法,它使用基因型数据来预测基因表达水平,并进一步关联这些表达变化与复杂性状。
汇总水平 TWAS
汇总水平的 TWAS 仅使用 GWAS 的汇总统计数据来进行表达预测和关联测试。这种方法通常涉及以下步骤:
-
汇总统计数据:从 GWAS 分析中获取汇总统计数据,这些数据通常是每个变异与性状关联的统计显著性。
-
表达预测:使用这些汇总统计数据来预测基因表达水平。这可能涉及到使用特定的算法,如 FUSION 或 S-PrediXcan,这些算法可以从 GWAS 结果中推断基因表达的变化。
-
关联测试:将预测的基因表达水平与性状数据进行关联测试,以识别可能影响性状的基因。
优势和挑战
- 优势:TWAS 可以提供关于基因如何通过调控表达来影响性状的直接证据,这对于理解遗传机制和疾病机理非常有用。
- 挑战:需要高质量的基因表达数据和遗传变异数据,以及复杂的统计分析方法。此外,对于汇总水平的 TWAS,预测的准确性可能受到限制,因为它们依赖于间接的推断。
总的来说,TWAS 是一种强大的工具,它结合了基因表达和遗传变异数据来识别与性状相关的基因,既可以在个体水平上进行,也可以在汇总水平上进行。
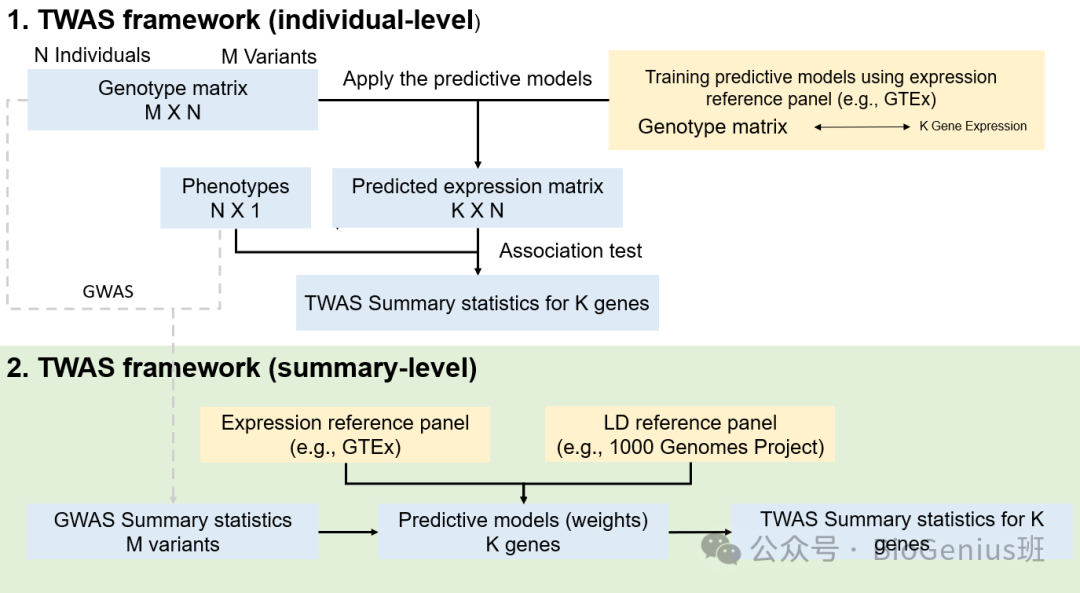
1. 个体水平的TWAS框架
- N 个体:研究中包含的个体数量。
- M 变异:基因组中考虑的遗传变异数量。
- 基因型矩阵(M x N):包含所有个体所有遗传变异的矩阵。
- 表型(N x 1):每个个体的表型数据。
- 预测表达矩阵(K x N):基于基因型矩阵预测的基因表达矩阵。
- 训练预测模型:使用表达参考面板(如GTEx,基因型-组织表达数据库)训练预测模型,将基因型矩阵转换为预测的表达矩阵。
- 关联测试:使用预测的表达矩阵和表型数据进行关联测试,以识别与表型显著相关的基因。
- TWAS 汇总统计:为K个基因生成的TWAS汇总统计数据。
2. 汇总水平的TWAS框架
- 表达参考面板(如GTEx):用于训练预测模型的基因表达数据集。
- LD参考面板(如1000基因组计划):用于训练预测模型的连锁不平衡(LD)参考数据集。
- GWAS 汇总统计(M 变异):从全基因组关联研究中获得的汇总统计数据。
- 预测模型(权重):使用表达参考面板和LD参考面板训练预测模型,为K个基因生成权重。
- TWAS 汇总统计:使用预测模型的权重和GWAS汇总统计生成K个基因的TWAS汇总统计数据。
关键步骤和概念
- 基因型矩阵:表示每个个体在每个遗传变异上的基因型。
- 表达矩阵:表示每个个体在每个基因上的表达水平。
- 预测模型:利用已知数据(如表达参考面板)来预测未知数据(如基因表达)的统计模型。
- 关联测试:统计方法,用于检测两个变量(如基因表达和表型)之间的关联。
- 汇总统计:在多个研究或多个变异中汇总得到的统计数据,用于简化和综合分析。
这两种TWAS框架的主要区别在于数据处理和分析的层次:个体水平框架直接处理个体的基因型和表达数据,而汇总水平框架则处理来自不同研究或数据集的汇总统计数据。汇总水平框架通常更容易实现,因为它不需要原始的基因表达数据,但可能需要高质量的汇总统计数据和参考面板。
4.FUSION http://gusevlab.org/projects/fusion/
工具介绍
- FUSION:这是一个用于执行转录组全关联研究的工具,它通过构建基因表达的遗传成分的预测模型,并使用全基因组关联研究(GWAS)的汇总统计数据来预测和测试这些成分与疾病的关联。
- 目标:FUSION的主要目标是识别全基因组关联研究(GWAS)中识别的性状与仅在参考数据中测量的功能性表型之间的关联。
使用方法
- 训练预测模型:FUSION通过使用表达参考面板(如GTEx,基因型-组织表达数据库)来训练预测模型,将基因型矩阵转换为预测的表达矩阵。
- 关联测试:使用预测的表达矩阵和表型数据进行关联测试,以识别与表型显著相关的基因。
参考文献
- 提到了一篇相关的科学论文,Gusev, A., Ko, A., Shi, H., Bhatia, G., Chung, W., Penninx, B. W., … & Pasaniuc, B. (2016). Integrative approaches for large-scale transcriptome-wide association studies. Nature Genetics, 48(3), 245-252,这篇论文详细介绍了FUSION方法和其在大规模转录组关联研究中的应用。
应用场景
- FUSION适用于那些需要在不直接测量基因表达水平的个体中,识别与复杂性状显著相关的基因的场景。
- 这种方法特别适用于效应量较小的表达-性状关联的检测,这些关联在传统的GWAS分析中可能难以被发现。
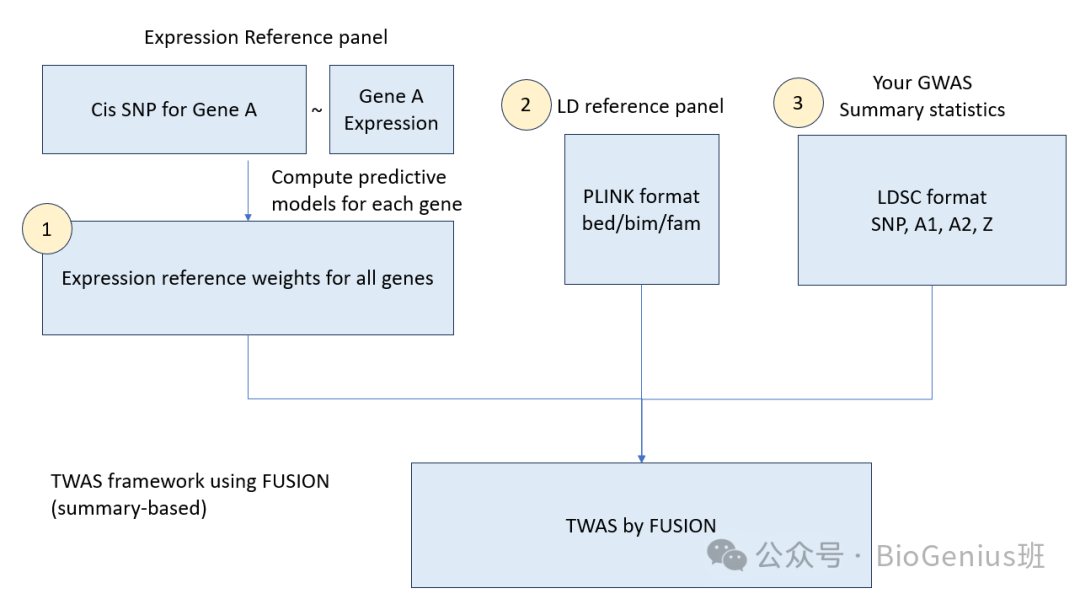
以下是图中各部分的详细解读:
1. 表达参考面板(Expression Reference Panel)
- Cis SNP for Gene A:表示与基因A相关的顺式SNP(cis-SNP),即基因内部的遗传变异,这些变异可能影响基因的表达水平。
- Gene A Expression:表示基因A的表达水平。
- Expression reference weights for all genes:通过计算每个基因的预测模型,得到所有基因的表达参考权重。这些权重用于预测基因表达水平。
2. 连锁不平衡参考面板(LD Reference Panel)
- PLINK format bed/bim/fam:连锁不平衡(LD)参考面板的数据格式,通常包含基因型数据,用于预测基因表达。
- 这些数据用于训练预测模型,将基因型转换为预测的表达矩阵。
3. 你的GWAS汇总统计(Your GWAS Summary Statistics)
- LDSC format SNP, A1, A2, Z:你的全基因组关联研究(GWAS)汇总统计数据,通常以LDSC格式提供,包括SNP值、等位基因频率(A1, A2)和统计显著性(Z值)。
TWAS by FUSION(TWAS Framework using FUSION)
- TWAS by FUSION:结合上述三个步骤,FUSION 软件包可以执行基于汇总数据的转录组全关联分析。它使用表达参考权重和GWAS汇总统计来预测和测试基因表达与性状之间的关联。